STRATEGIES IN STRUCTURE DETERMINATIONS
FROM POWDER DATA
2.2.2- "Automatic" indexing
Maybe your teacher said you that a simple slide rule will suffice for
indexing a powder pattern. That's true only if the problem is trivial.
Serious problems need a computer and telling that it is "automatic" is
truly a lie. You, in person, will have to make the final choice, frequently
among multiple propositions. Figures of merit will help you in your choice
and also your common sense. You should never have any full confidence in
a cell proposed by an indexing program. The ultimate proof that the cell
is correct is obtained when the structure has been determined and refined.
Even at this point, you may realize that the cell is in fact larger with
a higher symmetry or that you were in a wrong space group and so on.
Don't try any indexing program before to be sure that your data are
reliable. Never use only one indexing program. The most recommended actually
which have proven their hability in dozens of structure determinations
from powder data are TREOR, ITO and DICVOL
(you can find elsewhere other indexing programs which were not listed in
the SDPD-D because this databank gathers
only successful experimental cases), each of them has qualities and deficiencies.
You will have to read their manual and to test them on the cases included
in the packages (see links for obtaining programs
in the introduction). Test the program on some powder data of your own,
corresponding to known materials, so that you will obtain some proof that
you are ready to fight with a really unknown material. Gain some experience.
Why these advices ? The indexing step is certainly one of the most difficult
!
The strategy applied to the samples in this scenario was generally as
explained below :
-
Option b- of chapter 2.2.1 was selected : expecting
to succeed in a calibration by using harmonics (prefer option a- if you
are not yet an expert :-).
-
Patterns were recorded in the range 5-130 or even up to 150° 2-theta
; 0.02° 2-theta step scan ; time being 20-40 seconds per point, corresponding
to a long week-end ; sample holder with vertical loading : this could be
the final pattern for publication in case of success.
-
The pattern is processed : the background is estimated and subtracted from
the pattern ; Kalpha-2 is eliminated ; in some case the data are smoothed
(not really recommended, however the estimated angular positions with or
without smoothing will be acceptable if the resolution is good enough and
the step scan is as low as 0.02° 2-theta) ; peak positions are hunted
by using an algorithm based on derivatives (using the EVA software from
Socabim distributed by Siemens), another possibility would be to fit peaks
or groups of peaks by using pseudo-Voigt or other profile shapes like split
Pearson VII (...). I have not observed that profile fitting is better than
a derivative method when there is severe reflection overlapping. To decide
how many peaks are in a group can be a serious problem !
-
The result of the above efforts is a list of angular values to which are
corresponding d(Å) values and observed intensities (A demo by using
the PowderX software is available).
-
According to the calibration technique retained in this scenario, harmonics
have to be found. For instance, if a reflection is observed at 6.13Å,
one should find harmonics at 6.13/2=3.065; 6.13/3=2.043; 6.13/4=1.5325...
You have understood the principle. One should proceed tentatively up to
find 3 series of harmonics if possible. Only one series could suffice if
you detect up to 5 or more harmonics in it. Pressing the sample on its
holder will favour the detection of harmonics if a preferred orientation
occurs.
-
Using a software for cell refinement (as ERACEL for instance) applied to
the fictitious cell corresponding to the harmonics (an orthorhombic fictitious
cell if 3 series of harmonics have been detected), the a, b, c parameters
are refined together with a zeropoint (this zeropoint corresponding to
a systematic error which could be due to the goniometer + an error associated
with the fact that the sample is not exactly in the diffracting plane).
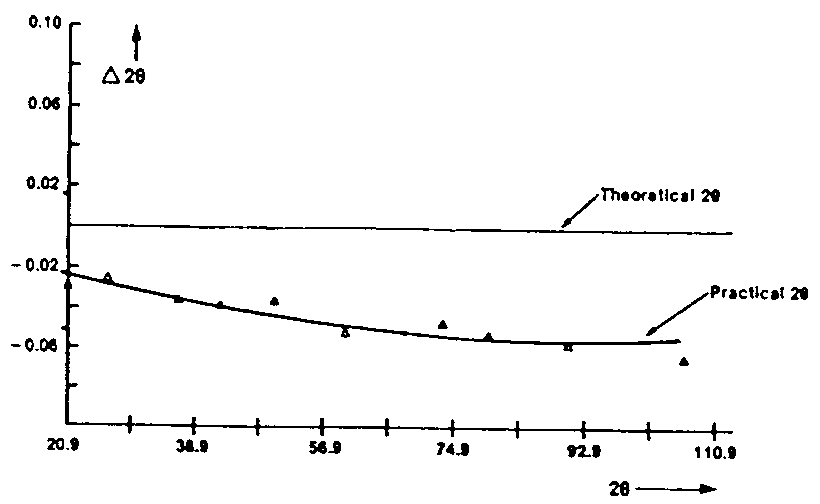
The "calibration" is finished. Really, the error cannot be reduced to a
zeropoint because the discrepancy between observed and theoretical angular
positions is not a constant and even does not vary linearly as a function
of the angle (see Fig. 10). However the
approximation by a constant zeropoint is sufficient for our first objective
which is indexing because we use generally data in the range 5-40°
2-theta where the error is almost a constant. Be careful, the maximum difference
between the observed and calculated angular positions corresponding to
your fictitious cell should be less than 0.01°(2-theta). If you have
a larger error, then eliminate the harmonic which appears clearly wrong
or eliminate a complete series of harmonics and replace it by another one.
Finally, if your result is not excellent, then calibrate your sample by
the first option : add a standard material to it and measure another pattern.
Taking the angular positions of the standard reflections, it may be sufficient
to refine a zeropoint which will be applied to the unknown. If really your
cell refinement gives discrepancies largest than 0.01°(2-theta), then
find another job :-).
-
Now is the indexing stage strictly speaking. You have to choose data, discarding
the dubious ones. In principle the 20 first reflections at low angle will
suffice. In this scenario, up to 3 programs will be tested (TREOR, ITO
and DICVOL). One should proceed slowly, realizing numerous tries enlarging
progressively the maximum cell volume, starting from, say 200Å3,
selecting first the highest symmetry and going progressively down the lowest
symmetries. The DICVOL program has no tolerance for unindexed lines. Personally,
I am usually uncertain of the sample purity. So that I tend to reserve
DICVOL for an ultimate confirmation and verification of the propositions
made by TREOR and ITO.
-
OK, and what to do now if a cell proposition seems convincing ? You should
use a program able to generate interreticular distances, choosing a P lattice
in the crystal system of the cell proposition. You have to convince yourself
evenmore that the cell is really correct and to have a space group proposition.
If you try to determine a structure from powder data with a wrong cell
or a wrong space group, then you will waste one or two weeks of your existence,
if not more. Thanks to this list of interreticular distances generated
by programs like LAZY-PULVERIX or ERADIS or even a Rietveld program able
to work in "calculated pattern" mode (like FULLPROF), one can index the
experimental data, noting possible extinctions. Doing this, the unambiguously
indexed reflections have to be selected in the objective to realize a definitive
cell parameters refinement. This final refinement should be perfect (errors
<0.01° 2-theta). The experts could avoid this unpleasing step, if
they are really sure of themselves, going directly to the next step. This
next step consists in an attempt to extract structure factors by the Pawley
method or the
Le Bail method.
The space group can be estimated at this next step too, realizing an extraction
in a space group without extinction (for instance choosing P2/m if the
system is monoclinic, P4/mmm if it is tetragonal...). Extracted intensities
have to be carefully checked in order to determine systematic extinctions
if any. The visual examination of a zoomed part of the pattern showing
the refinement result is essential for concluding to the absence or presence
of a reflection. If the cell was false, this step of extracting structure
factors will reveal it by a very bad correspondence between the observed
and cell-constrained calculated patterns.
We will see the results of this general strategy as applied to the experimental
cases retained in this scenario (warning : read
the manuals of the programs mentioned, they will not be fully restored
here) :
-/0/+
Copyright © 1997- Armel
Le Bail
{kind=link}