SDPD Internet Course
Semaine 10
Achèvement
de la détermination de structure et
affinement par la méthode
de Rietveld, niveau 2.
Lectures
Quand et comment arrêter un affinement de structure par la méthode de Rietveld est une question difficile, à laquelle vous ne pourrez vraiment répondre qu'après des années d'expérience. Devez-vous affiner les agitations thermiques de façon anisotrope parfois, au moins pour les atomes les plus lourds ? Devez-vous essayer de localiser les atomes d'hydrogène et les inclure dans l'affinement ? Quand donc les restrictions et contraintes sont-elles applicables ? Quel est le bon rapport minimum entre nombre de paramètres et de réflexions ? Est-ce que les paramètres de taille des cristallites et de distorsion du réseau, avec effet anisotrope possible, sont crédibles ou pas ? Etc. L'acceptation ou bien le rejet de votre manuscrit pour publication par un journal scientifique va dépendre de vos bons choix. Vous ne devez pas aller trop au-delà des limites du raisonnable, et ces limites sont certainement difficiles à établir exactement...
L'orientation préférentielle et l'élargissement anisotrope des pics de diffraction sont les deux principaux problèmes (si l'échantillon est pur, si la résolution est la meilleure possible et si la statistique est suffisante) qui peuvent limiter la qualité de vos affinements, et peuvent aussi vous empêcher de déterminer une structure par la méthode des poudres.
L'extraction de paramètres physiques, caractérisant les effets de taille des cristallites et de distorsion du réseau cristallin, à partir d'un diagramme de poudre, en possible en utilisant la methode de Rietveld. Mais c'est une approche controversée qui demande cette fois encore beaucoup d'expérience pour ne pas conclure n'importe quoi et être capable de répondre aux commentaires désagréables des referee. Deux livres de référence sur la question ont été publiés récemment :
B6- Defects and Microstructure Analysis
by Diffraction,
R.L. Snyder, J. Fiala and H.J. Bunge,
IUCr Monographs on Crystallography, Vol
10,
Oxford Science Publications, 1999.
B9 - Fundamentals of Powder Diffraction
and Structural Characterization of Materials
Vitalij K. Pecharsky, Peter Y. Zavalij
Kluwer Academic Pub., 2003.
Plusieurs chapitres concernent la méthode
de Rietveld. Vous aurez un aperçu des vues les plus récentes
en matière d'analyse de microstructure au moyen de la méthode
de Rietveld en lisant le texte (en anglais) d'une conférence donnée
à la "Sixth International School and Workshop of Crystallography",
22-27 janvier 2000, Ismailia, Egypte. Le Size/Strain Round
Robin voir l'article, voir aussi
une contribution)
est naturellement à examiner de très près (cependant,
le cas présenté est trop simple : cubique, isotrope, effet
de taile des cristallites seulement). Regardez aussi une application (format PDF) de la méthode
de Rietveld à une phase orthorhombique, avec des effects de taille
et de distorsion anisotropes. Voyez comment ces problèmes sont
traités dans Fullprof (CD: PDF).
Logiciels à télécharger
Vous pouvez rencontrer des problèmes particuliers qui vous empêchent de présenter des résultats complets tant que ces problèmes ne sont pas résolus: par exemple, ne pas localiser les atomes d'hydrogène pour un composé organique ou organométallique peut affecter vos reliabilité de plus de 5% sur les facteurs de reliabilité Rp, Rwp et RB. Mais vous pouvez avoir de sérieuses difficultés à localiser ces atomes d'hydrogène par une appproche de type Fourier différence. même ayant de très bonnes données synchrotron (il n'y aurait par contre pas de problème à localiser des atomes de deutérium en diffraction de neutrons - mais il faut deutérer l'échantillon). Une possibilité est de deviner la position des atomes d'hydrogène à l'aide d'un logiciel (SHELX, BABEL, etc) et d'affiner leurs positions sous contrainte. A part SHELX, les programmes qui peuvent proposer des positions pour les atomes d'hydrogène sont :
Babel, un logiciel utile pour des échanges de formats de données de coordonnées pour des structures organiques/organométalliques, et pour localiser des atomes d'hydrogène. Il existe une version avec une interface graphique pour MS Windows (babelwin.zip);
Xhydex pour localiser des atomes d'hydrogène dans des composés particuliers (hydrures) ;
Mol2Mol peut localiser des atomes d'hydrogène (logiciel commercial) ;
Molden semble en être capable également ;
WebLab visualiseur d'Accelrys/MSII (WLViewerLite40.exe);
VMD ;
Gromacs ;
Molden ;
SHELXL, déjà prrésenté.
Etc.
Tous ces programmes ne sont pas 100% efficaces, comme on peut le voir ci-dessous pour une application de BABEL à la tetracycline hydrochloride (SDPDRR) :
Position des atomes d'hydrogène,
tels qu'ils ont été localisés à partir des données
de diffraction synchrotron sur monocristal :
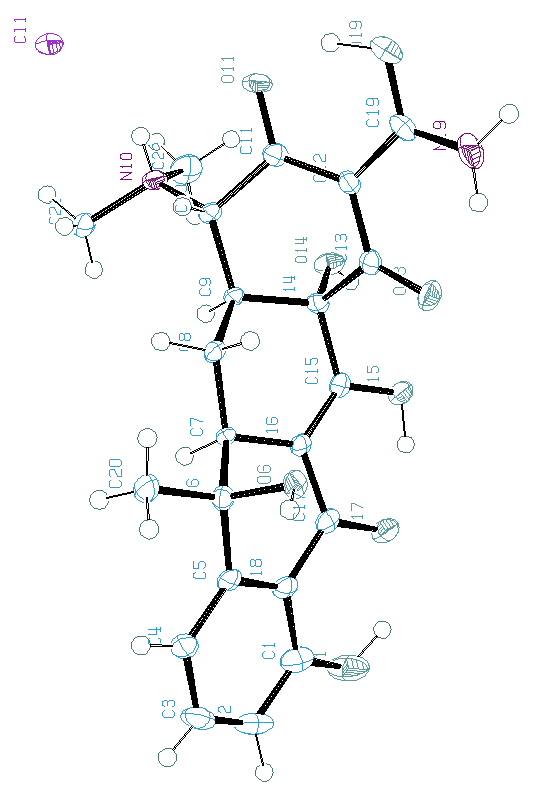
hydrogènes tels qu'ils sont
proposés par BABEL :
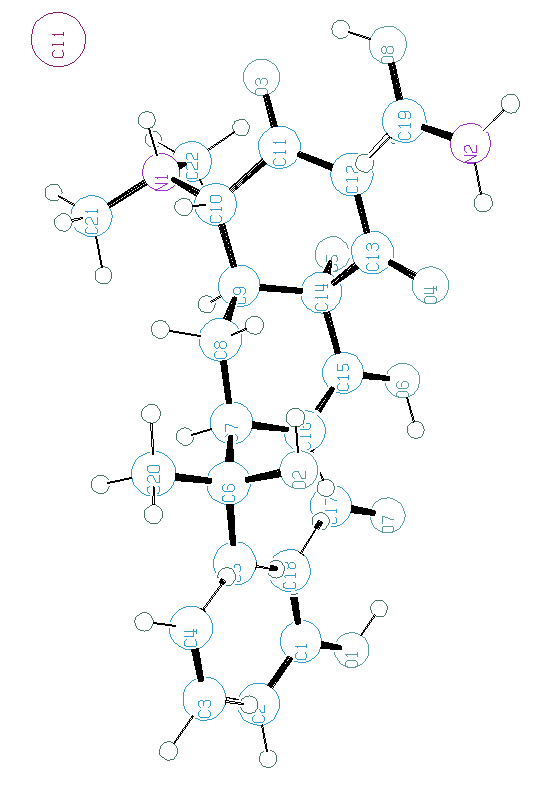
L'optimisation par méthode ab initio ou semi-empirique du modèle de molécule pourrait être fort utile avant d'affiner par la méthode de Rietveld C'est un peu hors sujet de ce cours, mais pensez-y. Des logiciels bien connus des chimistes peuvent le faire (SPARTAN, GAUSS...).
A nouveau à propos de microstructures,
de nombreux logiciels de type Rietveld autorisent cette approche. C'est un
domaine où vous devrez prendre vos responsabilités, parce la
discipline est à la limite de la connaissance reconnue. Parmi les
logiciels utiles dans ce domaine, on peut citer GSAS, FULLPROF, déjà
introduits la semaine précédente ou même avant, et WinMProf (qui
possède les plus récentes options de prise en compte des effets
d'anisotropie des réflexions - InstallWMP.exe).
Exercices
1- Complétez la détermination de structure du dérivé du pyrène (voyez la semaine 8). Essayez de localiser les atomes d'hydrogène. Ensuite, produisez le meilleur affinement que vous pourrez. Décidez si vous devez utiliser ou non des contraintes.
2- Essayez de faire de votre mieux avec la méthode de Rietveld appliquée au diagramme de diffraction synchrotron de la Norbornane (norbornane.dat, ou norbornane.pds, données prètes pour WinMProf) C17H12 at 50K (A.N. Fitch and H. Jobic, J. Chem. Soc. Chem. Commun. (1993) 1516-1517), en partant des coordonnées atomiques de la base de données de Cambridge (CSD) (norbor.txt, norbor.ps). Longueur d'onde 1.39839 A. Quels sont les problèmes principaux de ce diagramme de poudre ?
Dans chaque cas, expliquez brièvement
votre stratégie et produisez le fichier .sum si vous avez utilisé
FULLPROF, ou un résumé des résultats d'affinement équivalent
si vous avez utilisé un autre logiciel (incluant tous les paramètres
affinés, leur déviations standard estimées et les facteurs
de reliabilité).
Principaux logiciels sélectionnés pour la correction
BABEL, FULLPROF, SHELXL, ORTEP,
WinMProf (pour l'exercice 2)...
Deux semaines seront nécessaires
pour l'examen final.
Ces deux semaines peuvent commencer à
la date de votre choix. Mais le résultat doit être envoyé
au plus tard à la date prévue une fois ces deux semaines commencées.
Demandez-nous votre nombre de points total
accumulés depuis le début du cours, si vous ne l'avez pas déjà.
Bonne chance !