1- Na5Cr3F14
We will start from the last refinement by SHELXL97, done Week-6 :
"Na5Cr3F14" ??? P21/n ATOM x y z sof U11 Cr1 0.00000 0.00000 0.00000 0.50000 0.00975 0.00000 0.00000 0.00000 0.00000 0.00000 0.00258 Cr2 0.00000 -0.50000 0.00000 0.50000 0.01708 0.00000 0.00000 0.00000 0.00000 0.00000 0.00270 Cr3 0.00000 0.00000 0.50000 0.50000 0.01520 0.00000 0.00000 0.00000 0.00000 0.00000 0.00266 Na1 0.25203 -0.29049 0.23079 1.00000 0.01711 0.01553 0.00058 0.00149 0.00112 0.00000 0.00272 Na2 0.24149 -0.23213 0.71832 1.00000 0.02599 0.01584 0.00068 0.00137 0.00159 0.00000 0.00304 Na3 0.00000 -0.50000 0.50000 0.50000 0.04671 0.00000 0.00000 0.00000 0.00000 0.00000 0.00771 F1 0.11133 -0.48292 -0.19121 1.00000 0.02815 0.02857 0.00164 0.00187 0.00242 0.00000 0.00431 F2 0.13946 0.17073 0.52688 1.00000 0.02398 0.02376 0.00127 0.00193 0.00185 0.00000 0.00346 F3 -0.11215 0.20536 0.53896 1.00000 0.02629 0.02344 0.00113 0.00190 0.00198 0.00000 0.00363 F4 0.03585 -0.24438 0.06251 1.00000 0.02297 0.02015 0.00108 0.00163 0.00181 0.00000 0.00355 F5 -0.12985 -0.44000 -0.17702 1.00000 0.01530 0.01919 0.00091 0.00165 0.00146 0.00000 0.00342 F6 0.17163 0.03878 -0.00998 1.00000 0.02986 0.02805 0.00144 0.00211 0.00229 0.00000 0.00366 F7 0.00411 0.07027 0.24910 1.00000 0.01968 0.02050 0.00098 0.00180 0.00165 0.00000 0.00372 Final Structure Factor Calculation for "Na5Cr3F14" ??? P21/n Total number of l.s. parameters = 41 Maximum vector length = 511 Memory required = 2708 / 21973 R1 = 0.2220 for 1091 Fo > 4sig(Fo) and 0.2267 for all 1127 data wR2 = 0.7684, GooF = S = 11.182, Restrained GooF = 11.182 for all dataFrom these coordinates, the FULLPROF file is built (na5.pcr) :
COMM Na5Cr3F14 ?? ! Files => DAT-file: na5, PCR-file: na5 !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 1 14 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.60 0.60 0.60 0.80 10.0000 0.0100 150.0000 0.000 0.000 ! ! 2Theta/TOF Background 10.000 447.800 11.850 418.910 13.250 394.140 19.040 352.870 22.050 334.300 26.690 315.730 32.480 299.220 47.550 288.900 61.230 274.460 73.510 253.820 90.430 222.870 111.990 204.290 140.260 208.420 150.000 214.610 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 1 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0911 141.00 0.0000 0.00 0.0063 121.00 0.000000 0.00 0 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 33.18 !------------------------------------------------------------------------------- Na5Cr3F14 ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 13 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/N <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR1 CR+3 0.00000 0.00000 0.00000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 0.00 0.00 CR2 CR+3 0.00000 -0.50000 0.00000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 0.00 0.00 CR3 CR+3 0.00000 0.00000 0.50000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 0.00 0.00 NA1 NA+1 0.25203 -0.29049 0.23079 2.00000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 NA2 NA+1 0.24149 -0.23213 0.71832 2.00000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 NA3 NA+1 0.00000 -0.50000 0.50000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 0.00 0.00 F1 F-1 0.11133 -0.48292 -0.19121 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 F2 F-1 0.13946 0.17073 0.52688 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 F3 F-1 -0.11215 0.20536 0.53896 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 F4 F-1 0.03585 -0.24438 0.06251 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 F5 F-1 -0.12985 -0.44000 -0.17702 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 F6 F-1 0.17163 0.03878 -0.00998 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 F7 F-1 0.00411 0.07027 0.24910 1.20000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.10000E-01 0.4782 0.0000 0.0000 0.0000 0.0000 0 11.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.00159 0.00198 0.00173 0.00310 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 131.00 0.00 0.00 0.00 ! a b c alpha beta gamma 10.509497 7.225223 7.271324 90.000000 90.674828 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.02956 0.00088 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00Note that all the parameters refined at the "|Fobs|" extraction stage are kept fixed. Only a scale factor is refined at this stage. The fit is already quite good :
==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13999 => Rp: 8.05 Rwp: 10.7 Rexp: 2.81 Chi2: 14.5 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 10.1 Rwp: 12.7 Rexp: 3.35 Chi2: 14.5 => Deviance: 0.195E+06 Dev* : 13.94 => DW-Stat.: 0.1695 DW-exp: 1.9478 => N-sigma of the GoF: 1126.128 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13291 => Rp: 8.10 Rwp: 10.8 Rexp: 2.76 Chi2: 15.2 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 10.0 Rwp: 12.7 Rexp: 3.27 Chi2: 15.2 => Deviance: 0.194E+06 Dev* : 14.61 => DW-Stat.: 0.1703 DW-exp: 1.9464 => N-sigma of the GoF: 1154.139 => Phase: 1 => Bragg R-factor: 8.83 Vol: 552.098( 0.000) Fract(%): 0.00( 0.00) => Rf-factor= 6.69 ATZ: 0.000 Brindley: 1.0000
Now, and because the fit is already that good, we may expect to refine all the atomic coordinates as well as the isotropic thermal parameters (in a first step, grouping same atom-type with one thermal parameter). But the fit will not always be that good at this stage if the structure model is not complete. In such a case, it is recommended to fix the profile parameters (U, V, W, cell parameters, zeropoint, etc) that were obtained at the structure factors extraction stage, up to obtain a structure model which lead tp reliability factors sufficiently small (<20%), otherwise, those profile parameters may prove to be unstable and the fit may diverge.
In this especially straightforward case, let us refine all parameters
together (44 parameters already) :
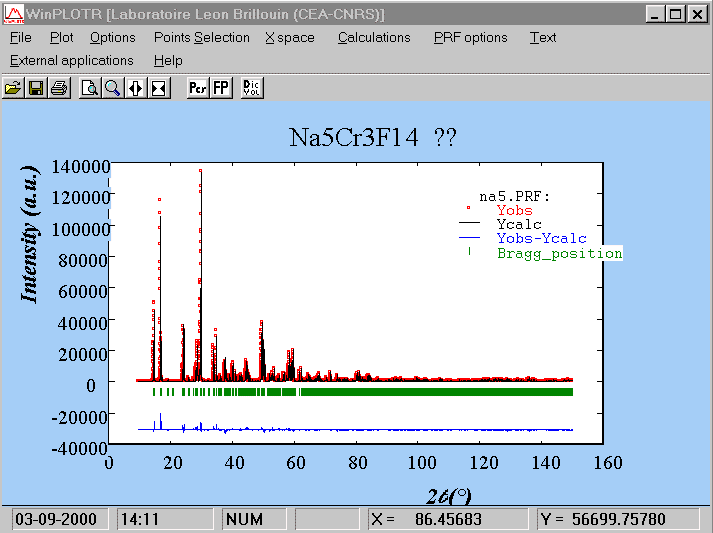
COMM Na5Cr3F14 ?? ! Files => DAT-file: na5, PCR-file: na5 !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 1 14 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.60 0.60 0.60 0.80 10.0000 0.0100 150.0000 0.000 0.000 ! ! 2Theta/TOF Background 10.000 447.800 11.850 418.910 13.250 394.140 19.040 352.870 22.050 334.300 26.690 315.730 32.480 299.220 47.550 288.900 61.230 274.460 73.510 253.820 90.430 222.870 111.990 204.290 140.260 208.420 150.000 214.610 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 44 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0911 141.00 0.0000 0.00 0.0063 121.00 0.000000 0.00 0 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 8.83 !------------------------------------------------------------------------------- Na5Cr3F14 ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 13 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/N <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR1 CR+3 0.00000 0.00000 0.00000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 CR2 CR+3 0.00000 -0.50000 0.00000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 CR3 CR+3 0.00000 0.00000 0.50000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 NA1 NA+1 0.25203 -0.29049 0.23079 2.00000 1.00000 0 0 0 151.00 161.00 171.00 431.00 0.00 NA2 NA+1 0.24149 -0.23213 0.71832 2.00000 1.00000 0 0 0 181.00 191.00 201.00 431.00 0.00 NA3 NA+1 0.00000 -0.50000 0.50000 0.50000 0.50000 0 0 0 0.00 0.00 0.00 431.00 0.00 F1 F-1 0.11133 -0.48292 -0.19121 1.20000 1.00000 0 0 0 211.00 221.00 231.00 441.00 0.00 F2 F-1 0.13946 0.17073 0.52688 1.20000 1.00000 0 0 0 241.00 251.00 261.00 441.00 0.00 F3 F-1 -0.11215 0.20536 0.53896 1.20000 1.00000 0 0 0 271.00 281.00 291.00 441.00 0.00 F4 F-1 0.03585 -0.24438 0.06251 1.20000 1.00000 0 0 0 301.00 311.00 321.00 441.00 0.00 F5 F-1 -0.12985 -0.44000 -0.17702 1.20000 1.00000 0 0 0 331.00 341.00 351.00 441.00 0.00 F6 F-1 0.17163 0.03878 -0.00998 1.20000 1.00000 0 0 0 361.00 371.00 381.00 441.00 0.00 F7 F-1 0.00411 0.07027 0.24910 1.20000 1.00000 0 0 0 391.00 401.00 411.00 441.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.52489E-02 0.4782 0.0000 0.0000 0.0000 0.0000 0 11.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.00159 0.00198 0.00173 0.00310 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 131.00 0.00 0.00 0.00 ! a b c alpha beta gamma 10.509497 7.225223 7.271324 90.000000 90.674843 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.02956 0.00088 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00The result is quite satisfying (.sum file) :
==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR1 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.904( 4) 0.500( 0) CR2 0.00000( 0) -0.50000( 0) 0.00000( 0) 0.904( 4) 0.500( 0) CR3 0.00000( 0) 0.00000( 0) 0.50000( 0) 0.904( 4) 0.500( 0) NA1 0.24828( 13) -0.29496( 14) 0.23245( 17) 2.393( 14) 1.000( 0) NA2 0.24434( 12) -0.23124( 15) 0.72045( 16) 2.393( 14) 1.000( 0) NA3 0.00000( 0) -0.50000( 0) 0.50000( 0) 0.893( 14) 0.500( 0) F1 0.11575( 12) -0.48895( 19) -0.19171( 18) 1.661( 11) 1.000( 0) F2 0.13435( 13) 0.16495( 18) 0.53026( 19) 1.661( 11) 1.000( 0) F3 -0.10988( 12) 0.20071( 18) 0.54342( 19) 1.661( 11) 1.000( 0) F4 0.03466( 13) -0.24671( 17) 0.06751( 19) 1.661( 11) 1.000( 0) F5 -0.12630( 13) -0.44057( 19) -0.17723( 18) 1.661( 11) 1.000( 0) F6 0.17573( 11) 0.03950( 18) -0.00907( 18) 1.661( 11) 1.000( 0) F7 0.00028( 14) 0.07217( 17) 0.24652( 20) 1.661( 11) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 10.50958 0.00002 7.22529 0.00001 7.27133 0.00001 90.00000 0.00000 90.67539 0.00011 90.00000 0.00000 => overall scale factor : 0.005705233 0.000006265 => Eta(p-v) or m(p-vii) : 0.43728 0.00400 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.00241 0.00006 0.00104 0.00006 0.00202 0.00002 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.06095 0.00201 0.00057 0.00032 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00437 0.00009 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0950 0.0002 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0032 0.0003 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13956 => Rp: 3.78 Rwp: 5.54 Rexp: 2.81 Chi2: 3.88 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 4.74 Rwp: 6.59 Rexp: 3.35 Chi2: 3.88 => Deviance: 0.542E+05 Dev* : 3.880 => DW-Stat.: 0.5371 DW-exp: 1.9539 => N-sigma of the GoF: 240.708 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13270 => Rp: 3.77 Rwp: 5.53 Rexp: 2.76 Chi2: 4.02 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 4.66 Rwp: 6.54 Rexp: 3.26 Chi2: 4.02 => Deviance: 0.532E+05 Dev* : 4.011 => DW-Stat.: 0.5461 DW-exp: 1.9529 => N-sigma of the GoF: 245.594 => Phase: 1 => Bragg R-factor: 3.58 Vol: 552.109( 0.002) Fract(%): 0.00( 0.00) => Rf-factor= 3.96 ATZ: 0.000 Brindley: 1.0000We may now try to do more. Instead of the manually estimated background, we may see if a polynomial background would not improve that fit. In such a case, the best is to put all 6 polynomial values to zero, excepted the first which can be given the value of the lowest background. Moreover, it is recommended to refine only the background values (note that if the pattern presents a limited 2-theta max value, it will not always be possible to refine all the 6 background polynomial values).
The new .pcr file becomes :
COMM Na5Cr3F14 ?? ! Files => DAT-file: na5, PCR-file: na5 !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 1 0 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.60 0.60 0.60 0.80 10.0000 0.0100 150.0000 0.000 0.000 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 6 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0950 141.00 0.0000 0.00 0.0032 121.00 0.000000 0.00 0 ! Background coefficients/codes 150.00 0.00 0.00 0.00 0.00 0.0 11.000 21.000 31.000 41.000 51.000 61.000 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 3.58 !------------------------------------------------------------------------------- Na5Cr3F14 ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 13 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/N <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR1 CR+3 0.00000 0.00000 0.00000 0.90367 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 CR2 CR+3 0.00000 -0.50000 0.00000 0.90367 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 CR3 CR+3 0.00000 0.00000 0.50000 0.90367 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 NA1 NA+1 0.24828 -0.29496 0.23245 2.39280 1.00000 0 0 0 151.00 161.00 171.00 431.00 0.00 NA2 NA+1 0.24434 -0.23124 0.72045 2.39280 1.00000 0 0 0 181.00 191.00 201.00 431.00 0.00 NA3 NA+1 0.00000 -0.50000 0.50000 0.89280 0.50000 0 0 0 0.00 0.00 0.00 431.00 0.00 F1 F-1 0.11575 -0.48895 -0.19171 1.66140 1.00000 0 0 0 211.00 221.00 231.00 441.00 0.00 F2 F-1 0.13435 0.16495 0.53026 1.66140 1.00000 0 0 0 241.00 251.00 261.00 441.00 0.00 F3 F-1 -0.10988 0.20071 0.54342 1.66140 1.00000 0 0 0 271.00 281.00 291.00 441.00 0.00 F4 F-1 0.03466 -0.24671 0.06751 1.66140 1.00000 0 0 0 301.00 311.00 321.00 441.00 0.00 F5 F-1 -0.12630 -0.44057 -0.17723 1.66140 1.00000 0 0 0 331.00 341.00 351.00 441.00 0.00 F6 F-1 0.17573 0.03950 -0.00907 1.66140 1.00000 0 0 0 361.00 371.00 381.00 441.00 0.00 F7 F-1 0.00028 0.07217 0.24652 1.66140 1.00000 0 0 0 391.00 401.00 411.00 441.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.57052E-02 0.4373 0.0000 0.0000 0.0000 0.0000 0 451.00000 461.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.00241 0.00104 0.00202 0.00437 0.00000 0.00000 0.00000 0 491.00 481.00 471.00 131.00 0.00 0.00 0.00 ! a b c alpha beta gamma 10.509584 7.225294 7.271334 90.000000 90.675392 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.06095 0.00057 0.00000 0.00000 0.00 0.00 501.00 71.00 0.00 0.00A first run is done with those 6 parameters only, and then, if the refinement is satisfying, the whole set of parameters are refined (now 50 parameters) :
=> No. of reflections: 2264 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR1 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.816( 5) 0.500( 0) CR2 0.00000( 0) -0.50000( 0) 0.00000( 0) 0.816( 5) 0.500( 0) CR3 0.00000( 0) 0.00000( 0) 0.50000( 0) 0.816( 5) 0.500( 0) NA1 0.24815( 12) -0.29537( 13) 0.23214( 16) 2.321( 14) 1.000( 0) NA2 0.24418( 11) -0.23082( 14) 0.71992( 15) 2.321( 14) 1.000( 0) NA3 0.00000( 0) -0.50000( 0) 0.50000( 0) 0.821( 14) 0.500( 0) F1 0.11566( 11) -0.48872( 18) -0.19181( 16) 1.607( 11) 1.000( 0) F2 0.13429( 12) 0.16474( 17) 0.53039( 18) 1.607( 11) 1.000( 0) F3 -0.10967( 11) 0.20043( 17) 0.54343( 18) 1.607( 11) 1.000( 0) F4 0.03485( 12) -0.24676( 16) 0.06737( 18) 1.607( 11) 1.000( 0) F5 -0.12601( 12) -0.44067( 17) -0.17694( 16) 1.607( 11) 1.000( 0) F6 0.17565( 10) 0.03977( 16) -0.00912( 17) 1.607( 11) 1.000( 0) F7 0.00002( 13) 0.07200( 16) 0.24582( 19) 1.607( 11) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 10.50958 0.00002 7.22528 0.00001 7.27133 0.00001 90.00000 0.00000 90.67535 0.00010 90.00000 0.00000 => overall scale factor : 0.005666310 0.000006379 => Eta(p-v) or m(p-vii) : 0.42389 0.00380 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.00255 0.00006 0.00090 0.00006 0.00205 0.00002 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.06045 0.00191 0.00056 0.00030 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00483 0.00008 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0950 0.0002 => Background Polynomial Parameters ==> 203.253 0.805318 -142.554 3.25243 404.226 9.36827 601.180 19.6487 -862.140 22.7986 -1367.21 33.1536 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0032 0.0003 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13950 => Rp: 3.60 Rwp: 5.19 Rexp: 2.81 Chi2: 3.41 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 4.48 Rwp: 6.15 Rexp: 3.33 Chi2: 3.41 => Deviance: 0.481E+05 Dev* : 3.448 => DW-Stat.: 0.6072 DW-exp: 1.9548 => N-sigma of the GoF: 201.555 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13264 => Rp: 3.59 Rwp: 5.18 Rexp: 2.76 Chi2: 3.53 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 4.40 Rwp: 6.09 Rexp: 3.24 Chi2: 3.53 => Deviance: 0.473E+05 Dev* : 3.562 => DW-Stat.: 0.6182 DW-exp: 1.9538 => N-sigma of the GoF: 205.711 => Phase: 1 => Bragg R-factor: 3.24 Vol: 552.108( 0.002) Fract(%): 0.00( 0.00) => Rf-factor= 3.11 ATZ: 0.000 Brindley: 1.0000
We can now try to refine all thermal parameters independently (60
parameters) :
COMM Na5Cr3F14 ?? ! Files => DAT-file: na5, PCR-file: na5 !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 1 0 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.60 0.60 0.60 0.80 10.0000 0.0100 150.0000 0.000 0.000 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 60 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0950 141.00 0.0000 0.00 0.0032 121.00 0.000000 0.00 0 ! Background coefficients/codes 203.25 -142.55 404.23 601.18 -862.14 -1367.2 11.000 21.000 31.000 41.000 51.000 61.000 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 3.24 !------------------------------------------------------------------------------- Na5Cr3F14 ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 13 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/N <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR1 CR+3 0.00000 0.00000 0.00000 0.81624 0.50000 0 0 0 0.00 0.00 0.00 421.00 0.00 CR2 CR+3 0.00000 -0.50000 0.00000 0.81624 0.50000 0 0 0 0.00 0.00 0.00 511.00 0.00 CR3 CR+3 0.00000 0.00000 0.50000 0.81624 0.50000 0 0 0 0.00 0.00 0.00 521.00 0.00 NA1 NA+1 0.24815 -0.29537 0.23214 2.32138 1.00000 0 0 0 151.00 161.00 171.00 431.00 0.00 NA2 NA+1 0.24418 -0.23082 0.71992 2.32138 1.00000 0 0 0 181.00 191.00 201.00 531.00 0.00 NA3 NA+1 0.00000 -0.50000 0.50000 0.82138 0.50000 0 0 0 0.00 0.00 0.00 541.00 0.00 F1 F-1 0.11566 -0.48872 -0.19181 1.60745 1.00000 0 0 0 211.00 221.00 231.00 441.00 0.00 F2 F-1 0.13429 0.16474 0.53039 1.60745 1.00000 0 0 0 241.00 251.00 261.00 551.00 0.00 F3 F-1 -0.10967 0.20043 0.54343 1.60745 1.00000 0 0 0 271.00 281.00 291.00 561.00 0.00 F4 F-1 0.03485 -0.24676 0.06737 1.60745 1.00000 0 0 0 301.00 311.00 321.00 571.00 0.00 F5 F-1 -0.12601 -0.44067 -0.17694 1.60745 1.00000 0 0 0 331.00 341.00 351.00 581.00 0.00 F6 F-1 0.17565 0.03977 -0.00912 1.60745 1.00000 0 0 0 361.00 371.00 381.00 591.00 0.00 F7 F-1 0.00002 0.07200 0.24582 1.60745 1.00000 0 0 0 391.00 401.00 411.00 601.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.56663E-02 0.4239 0.0000 0.0000 0.0000 0.0000 0 451.00000 461.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.00255 0.00090 0.00205 0.00483 0.00000 0.00000 0.00000 0 491.00 481.00 471.00 131.00 0.00 0.00 0.00 ! a b c alpha beta gamma 10.509578 7.225283 7.271332 90.000000 90.675354 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.06045 0.00056 0.00000 0.00000 0.00 0.00 501.00 71.00 0.00 0.00The final result is shown in the following .sul FULLPROF file :
******************************************************** ** PROGRAM FULLPROF.98 (Version 0.2 - Mar98-LLB JRC) ** ******************************************************** Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 09/03/2000 Time: 15:21:41.620 => PCR file code: na5 => DAT file code: na5 => Title: Na5Cr3F14 ?? ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 => Base of peaks: 2.0*HW* 15.00 ==> Angular range, step and number of points: 2Thmin: 10.0000 2Thmax: 150.0000 Step: 0.0100 No. of points: 14001 => Crystal Structure Refinement for phase: 1 => Scor: 1.4856 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 60 ------------------------------------------------------------------------------ => Phase No. 1 Na5Cr3F14 ?? P 21/N ------------------------------------------------------------------------------ => No. of reflections: 2264 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR1 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.474( 6) 0.500( 0) CR2 0.00000( 0) -0.50000( 0) 0.00000( 0) 0.797( 8) 0.500( 0) CR3 0.00000( 0) 0.00000( 0) 0.50000( 0) 0.718( 7) 0.500( 0) NA1 0.24793( 6) -0.29532( 7) 0.23174( 8) 1.155( 12) 1.000( 0) NA2 0.24354( 6) -0.23092( 8) 0.71919( 9) 2.495( 16) 1.000( 0) NA3 0.00000( 0) -0.50000( 0) 0.50000( 0) 3.140( 21) 0.500( 0) F1 0.11488( 6) -0.48831( 10) -0.19280( 9) 1.798( 18) 1.000( 0) F2 0.13435( 7) 0.16452( 10) 0.52962( 10) 1.844( 21) 1.000( 0) F3 -0.10973( 6) 0.19977( 10) 0.54397( 10) 1.593( 19) 1.000( 0) F4 0.03450( 7) -0.24697( 9) 0.06798( 10) 1.588( 17) 1.000( 0) F5 -0.12715( 7) -0.44018( 10) -0.17783( 9) 1.552( 19) 1.000( 0) F6 0.17580( 6) 0.04025( 10) -0.00967( 10) 2.056( 19) 1.000( 0) F7 -0.00003( 8) 0.07193( 9) 0.24676( 11) 2.182( 18) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 10.50959 0.00001 7.22529 0.00001 7.27133 0.00001 90.00000 0.00000 90.67531 0.00006 90.00000 0.00000 => overall scale factor : 0.005670784 0.000003652 => Eta(p-v) or m(p-vii) : 0.42776 0.00217 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.00260 0.00004 0.00089 0.00003 0.00203 0.00001 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.05628 0.00114 0.00045 0.00017 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00466 0.00005 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0945 0.0001 => Background Polynomial Parameters ==> 189.152 0.480015 -167.773 1.88409 449.249 5.51788 662.940 11.3522 -935.337 13.4736 -1447.58 19.3529 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0036 0.0001 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13940 => Rp: 1.86 Rwp: 2.98 Rexp: 2.81 Chi2: 1.13 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 2.29 Rwp: 3.51 Rexp: 3.31 Chi2: 1.13 => Deviance: 0.158E+05 Dev* : 1.133 => DW-Stat.: 1.7860 DW-exp: 1.9562 => N-sigma of the GoF: 10.505 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13251 => Rp: 1.82 Rwp: 2.92 Rexp: 2.76 Chi2: 1.12 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 2.21 Rwp: 3.41 Rexp: 3.23 Chi2: 1.12 => Deviance: 0.149E+05 Dev* : 1.127 => DW-Stat.: 1.8890 DW-exp: 1.9553 => N-sigma of the GoF: 9.755 => Phase: 1 => Bragg R-factor: 0.456 Vol: 552.109( 0.001) Fract(%): 0.00( 0.00) => Rf-factor=0.509 ATZ: 0.000 Brindley: 1.0000
This is too good ! You will have some explanation Week-10 why this is too good. In standard cases, you may expect Rp in the range 7-12% for a quite good fit, and the Bragg R factor in the range 3-5%. Larger R values are of course possible but would be indicative of some difficulties. Smaller values are possible but quite rare.
No interest here in trying to refine anisotropic thermal parameters. We do not mean that this is always nonsense. From neutron data on PbSO4, this would probably feasible (see the RRRR - Rietveld Refinement Round Robin), or from X-ray data on the Pb atom only.
1- Pb2CrF7
We will do the job as if only Pb and Cr atoms had been located before Rietveld refinement.
At week-6 the coordinates proposed by the SHELXS-97 direct methods,
and then refined by using SHELXL-97, were :
"PbF2/CrF3/HFaq" ??? P21/c ATOM x y z sof U Pb1 0.21861 0.18445 0.05435 1.00000 0.01683 0.01290 0.00133 0.00076 0.00082 0.00000 0.00456 Pb2 0.24052 0.56295 0.13631 1.00000 0.01793 0.01070 0.00125 0.00052 0.00063 0.00000 0.00468 Cr1 0.28704 0.87394 0.20097 1.00000 0.01141 0.03835 0.00490 0.00204 0.00273 0.00000 0.01016 R1 = 0.2479 for 261 Fo > 4sig(Fo) and 0.2505 for all 270 data wR2 = 0.7832, GooF = S = 11.707, Restrained GooF = 11.707 for all dataWe have to build the FULLPROF .pcr file, using the previous file built for structure factors extraction (week-5). Of course, we should use the pattern where the contribution of the CrF3 impurity is the smallest. This time again, we will only refine the scale factor of the main phase (pbcr.pcr) :
COMM PbCr ?? ! Files => DAT-file: pbcr, PCR-file: pbcr !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 2 11 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.60 0.60 0.60 0.80 5.0000 0.0100 150.0000 0.000 0.000 ! ! 2Theta/TOF Background 5.000 530.760 6.920 489.660 9.080 446.920 13.880 389.390 16.760 358.160 22.040 333.500 27.570 312.130 39.810 287.470 84.940 256.240 112.070 246.380 150.000 267.750 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 1 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0337 121.00 0.0000 0.00 0.0000 0.00 0.000000 0.00 0 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 1.82 !------------------------------------------------------------------------------- PbCr ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 3 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/C <--Space group symbol PB1 PB+2 0.21861 0.18445 0.05435 1.00000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 PB2 PB+2 0.24052 0.56295 0.13631 1.00000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 CR CR+3 0.28704 0.87394 0.20097 0.50000 1.00000 0 0 0 0.00 0.00 0.00 0.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.14573E-03 0.7855 0.0000 0.0000 0.0000 0.0000 0 11.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.01160 0.00031 0.00182 0.00000 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 0.00 0.00 0.00 0.00 ! a b c alpha beta gamma 5.462546 11.208424 9.573616 90.000000 91.196121 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.02525 0.00100 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00 !------------------------------------------------------------------------------- ! Data for PHASE number: 2 ==> Current R_Bragg: 1.63 !------------------------------------------------------------------------------- CrF3 ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 2 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! R -3 C <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR CR+3 0.00000 0.00000 0.00000 0.50000 0.06000 0 0 0 0.00 0.00 0.00 0.00 0.00 F F-1 0.60000 0.00000 0.25000 0.80000 0.18000 0 0 0 0.00 0.00 0.00 0.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.14573E-01 0.7855 0.0000 0.0000 0.0000 0.0000 0 151.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.01160 0.00031 0.00182 0.00000 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 0.00 0.00 0.00 0.00 ! a b c alpha beta gamma 4.976699 4.976699 13.144844 90.000000 90.000000 120.000000 131.00000 131.00000 141.00000 0.00000 0.00000 131.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.02525 0.00100 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00
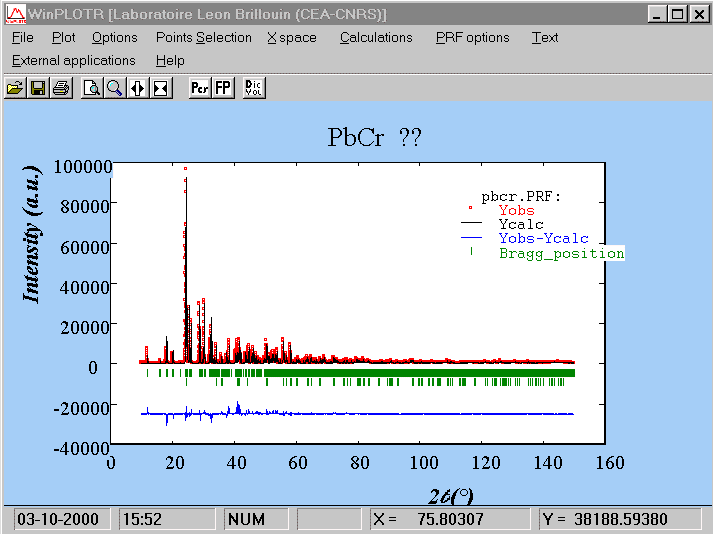
The fit is already impressive :
And the reliability factors are much smaller than those produced by
the SHELXL-97 refinement using the extracted structure factors. In principle
R1 from SHELX can be compared to RF from the Rietveld method. We had R1
= 25 % and we have RF = 7.3 %, as seen in the FULLPROF .sum file, without
refining nothing but the scale factor. This gives an idea of the weak contribution
of the F atoms in that structure :
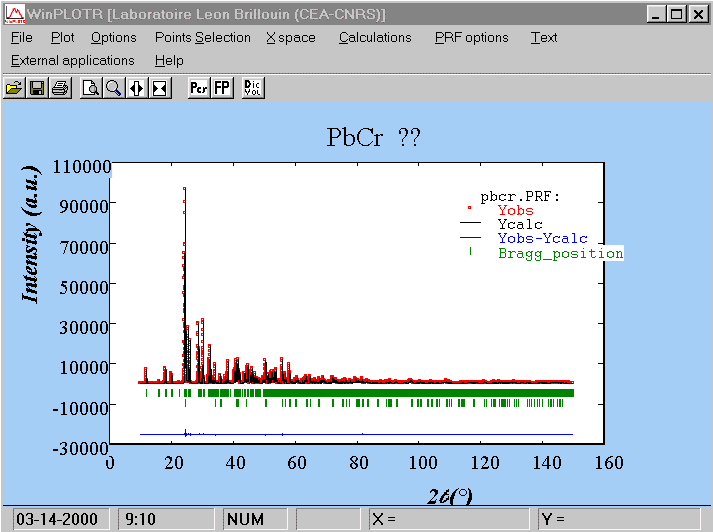
******************************************************** ** PROGRAM FULLPROF.98 (Version 0.2 - Mar98-LLB JRC) ** ******************************************************** Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 10/03/2000 Time: 16:07:52.220 => PCR file code: pbcr => DAT file code: pbcr => Title: PbCr ?? ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 => Base of peaks: 2.0*HW* 15.00 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 150.0000 Step: 0.0100 No. of points: 14501 => Crystal Structure Refinement for phase: 1 => Crystal Structure Refinement for phase: 2 => Scor: 7.6344 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 1 ------------------------------------------------------------------------------ => Phase No. 1 PbCr ?? P 21/C ------------------------------------------------------------------------------ => No. of reflections: 2403 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. PB1 0.21861( 0) 0.18445( 0) 0.05435( 0) 1.000( 0) 1.000( 0) PB2 0.24052( 0) 0.56295( 0) 0.13631( 0) 1.000( 0) 1.000( 0) CR 0.28704( 0) 0.87394( 0) 0.20097( 0) 0.500( 0) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 5.46255 0.00000 11.20842 0.00000 9.57362 0.00000 90.00000 0.00000 91.19613 0.00000 90.00000 0.00000 => overall scale factor : 0.000265872 0.000000575 => Eta(p-v) or m(p-vii) : 0.78550 0.00000 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.01160 0.00000 0.00031 0.00000 0.00182 0.00000 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.02525 0.00000 0.00100 0.00000 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ------------------------------------------------------------------------------ => Phase No. 2 CrF3 R -3 C ------------------------------------------------------------------------------ => No. of reflections: 140 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.500( 0) 0.060( 0) F 0.60000( 0) 0.00000( 0) 0.25000( 0) 0.800( 0) 0.180( 0) ==> PROFILE PARAMETERS: => Cell parameters : 4.97670 0.00000 4.97670 0.00000 13.14484 0.00000 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => overall scale factor : 0.014573000 0.000000000 => Eta(p-v) or m(p-vii) : 0.78550 0.00000 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.01160 0.00000 0.00031 0.00000 0.00182 0.00000 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.02525 0.00000 0.00100 0.00000 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0337 0.0000 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 2 => MaxCycle: 10 => N-P+C: 13998 => Rp: 11.6 Rwp: 16.5 Rexp: 3.05 Chi2: 29.4 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 15.6 Rwp: 21.1 Rexp: 3.88 Chi2: 29.4 => Deviance: 0.465E+06 Dev* : 33.21 => DW-Stat.: 0.1032 DW-exp: 1.9478 => N-sigma of the GoF: 2377.642 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13361 => Rp: 11.7 Rwp: 16.7 Rexp: 3.00 Chi2: 30.8 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 15.5 Rwp: 21.0 Rexp: 3.79 Chi2: 30.8 => Deviance: 0.464E+06 Dev* : 34.73 => DW-Stat.: 0.1034 DW-exp: 1.9465 => N-sigma of the GoF: 2432.603 => Phase: 1 => Bragg R-factor: 14.5 Vol: 586.031( 0.000) Fract(%): 0.00( 0.00) => Rf-factor= 7.27 ATZ: 0.000 Brindley: 1.0000 => Phase: 2 => Bragg R-factor: 5.79 Vol: 281.948( 0.000) Fract(%): 0.00( 0.00) => Rf-factor= 4.40 ATZ: 0.000 Brindley: 1.0000 => Run finished at: Date: 10/03/2000 Time: 16:08:16.060With such a good result, we can now refine all parameters, including two thermal parameters (one for the Cr atom and one for the two Pb atoms). That step is realized with the fp90.exe program, in order to obtain a correct .fou file (fp98 is a beta version with some bugs (? from our point of view, we don't know if this is not the author's desire ) at this step, sometimes). The .sum file is below, showing that a few % were gained :
******************************************************* *** PROGRAM FULLPROF (Version 3.3 - Aug97-LLB JRC) *** ******************************************************* Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 03/13/00 Time: 15:50:44.84 => PCR file code: pbcr => DAT file code: pbcr => Title: PbCr ?? ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Base of peaks: 2.0*HW* 15.00 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 150.0000 Step: 0.0100 No. of points: 14501 => Crystal Structure Refinement for phase: 1 => Crystal Structure Refinement for phase: 2 => Scor: 7.0213 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 27 ------------------------------------------------------------------------------ => Phase No. 1 PbCr ?? P 21/C ------------------------------------------------------------------------------ => No. of reflections: 2403 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. PB1 0.21935( 28) 0.18913( 14) 0.05528( 16) 1.029( 29) 1.000( 0) PB2 0.23843( 29) 0.56035( 15) 0.13402( 15) 1.240( 32) 1.000( 0) CR 0.29400( 101) 0.86764( 51) 0.20427( 58) 0.316(120) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 5.46354 0.00005 11.21106 0.00011 9.57593 0.00010 90.00000 0.00000 91.19402 0.00069 90.00000 0.00000 => Overall scale factor : 0.000271658 0.000001122 => ETA(p-V) or M(P-VII) : 0.64675 0.00762 => Overall Tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.02743 0.00087 -0.01386 0.00076 0.00608 0.00016 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.15855 0.00307 -0.01270 0.00107 0.00000 0.00000 0.00000 0.00000 => X and Y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ------------------------------------------------------------------------------ => Phase No. 2 CrF3 R -3 C ------------------------------------------------------------------------------ => No. of reflections: 140 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.500( 0) 0.060( 0) F 0.60000( 0) 0.00000( 0) 0.25000( 0) 0.800( 0) 0.180( 0) ==> PROFILE PARAMETERS: => Cell parameters : 4.97771 0.00005 4.97771 0.00005 13.14706 0.00022 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => Overall scale factor : 0.015101586 0.000085291 => ETA(p-V) or M(P-VII) : 0.64675 0.00762 => Overall Tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.02743 0.00087 -0.01386 0.00076 0.00608 0.00016 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.15855 0.00307 -0.01270 0.00107 0.00000 0.00000 0.00000 0.00000 => X and Y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0531 0.0004 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13972 => Rp: 11.0 Rwp: 15.6 Rexp: 3.05 Chi2: 26.4 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 14.9 Rwp: 19.9 Rexp: 3.88 Chi2: 26.4 => Deviance: 0.416E+06 Dev* : 29.76 => DW-Stat.: 0.1389 DW-exp: 1.9515 => N-sigma of the GoF: 2120.825 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13605 => Rp: 11.1 Rwp: 15.7 Rexp: 3.02 Chi2: 27.1 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 14.8 Rwp: 19.9 Rexp: 3.83 Chi2: 27.1 => Deviance: 0.416E+06 Dev* : 30.54 => DW-Stat.: 0.1390 DW-exp: 1.9509 => N-sigma of the GoF: 2149.244 => Phase: 1 => Bragg R-factor: 12.9 Vol: 586.419 Fract(%): 0.00 => Rf-factor= 6.18 ATZ: 0.000 Brindley: 1.0000 => Phase: 2 => Bragg R-factor: 4.52 Vol: 282.110 Fract(%): 0.00 => Rf-factor= 3.59 ATZ: 0.000 Brindley: 1.0000A Fourier synthesis will be calculated by the SHELXL-97 program, from the "|Fobs|" produced by the Rietveld decomposition formula (pbcr.fou). At the end of this document, another approach will be proposed, by using the GFOUR program. Remember that the "|Fobs|" depends on the |Fcalc| in that formula, so that a Fourier synthesis is less efficient than from single crystal data. Moreover, in that normal use of the Rietveld decomposition formula, only one iteration is performed, contrarily to what happens in the Le Bail method for "|Fobs|" extraction. In a first aproach, we will not use the OVERLAP program, and try the whole dataset. Below is the SHELXL .ins file :
TITL "PbF2/CrF3/HFaq" ??? P21/c CELL 1.54056 5.46255 11.20846 9.57362 90.0 91.19608 90.0 ZERR 2 0.001 0.001 0.001 0. 0.001 0. LATT 1 SYMM X,0.5-Y,0.5+Z SFAC PB CR F UNIT 8 4 28 L.S. 4 BOND ACTA FMAP 2 PLAN -10 FVAR 1 PB1 1 0.21861 0.18445 0.05435 11.00000 0.01266 PB2 1 0.24052 0.56295 0.13631 11.00000 0.01266 CR 2 0.28704 0.87394 0.20097 11.00000 0.00633 HKLF 3 END
We retrieve the R1 value equivalent to the Rietveld RB value after
the SHELXL refinement :
"PbF2/CrF3/HFaq" ??? P21/c ATOM x y z sof U11 U22 U33 U23 U13 U12 Ueq Pb1 0.21949 0.18912 0.05528 1.00000 0.01534 0.00046 0.00005 0.00002 0.00003 0.00000 0.00040 Pb2 0.23830 0.56038 0.13397 1.00000 0.01790 0.00054 0.00005 0.00002 0.00003 0.00000 0.00040 Cr 0.29464 0.86749 0.20458 1.00000 0.00546 0.00194 0.00017 0.00008 0.00013 0.00000 0.00044 Final Structure Factor Calculation for "PbF2/CrF3/HFaq" ??? P21/c Total number of l.s. parameters = 13 Maximum vector length = 511 Memory required = 420 / 21973 wR2 = 0.1822 before cycle 5 for 1206 data and 0 / 13 parameters GooF = S = 0.242; Restrained GooF = 0.242 for 0 restraints Weight = 1 / [ sigma^2(Fo^2) + ( 0.1000 * P )^2 + 0.00 * P ] where P = ( Max ( Fo^2, 0 ) + 2 * Fc^2 ) / 3 R1 = 0.0737 for 82 Fo > 4sig(Fo) and 0.0612 for all 1206 data wR2 = 0.1822, GooF = S = 0.242, Restrained GooF = 0.242 for all dataAn we look at the Fourier difference in the pbcr.lst file :
Atom Peak x y z Sof Height Distances and Angles PB1 2.12 0.2195 0.1891 0.0553 1.000 3.3 0 PB2 4.230 0 8 1.984 94.3 0 5 2.199 28.8 90.6 0 7 2.274 25.6 78.4 14.8 116 10 2.404 87.7 90.9 116.4 107.7 0 2 2.446 91.6 50.9 67.5 66.0 141.7 79 6 2.576 66.3 30.1 71.6 57.0 79.3 65.5 0 9 2.595 137.0 48.4 138.7 126.7 74.6 80.2 72.0 0 1 2.851 81.9 158.9 76.1 90.2 109.6 108.3 146.8 140.8 31 1 2.968 69.3 86.7 97.6 88.5 19.2 132.9 67.4 85.7 110.8 48 3 2.971 134.6 129.1 124.3 139.0 102.1 105.3 158.8 87.8 53.0 118.9 37 2 3.162 89.2 155.1 103.8 112.0 64.7 153.7 137.4 116.0 45.9 71.5 0 3 3.197 133.7 99.6 161.4 156.2 48.4 130.8 110.5 54.5 98.0 67.7 0 4 3.222 42.0 126.4 64.5 67.0 64.3 132.0 96.5 138.8 62.4 54.0 0 6 3.301 88.8 99.6 61.2 72.1 169.2 48.6 108.5 114.5 59.8 157.7 106 9 3.449 136.7 85.3 107.9 114.6 135.6 55.2 113.0 70.1 83.5 153.3 0 10 3.655 71.9 149.3 64.0 78.2 115.1 101.0 135.2 151.0 12.2 112.2 22 CR 3.730 105.7 159.5 104.8 118.6 94.3 122.6 169.8 114.2 28.8 104.3 23 CR 3.767 107.4 75.9 78.7 84.0 160.4 29.7 95.1 85.9 85.4 162.1 20 CR 3.897 147.4 73.8 164.2 150.2 62.9 102.8 93.2 26.1 119.5 79.7 PB2 1.79 0.2383 0.5604 0.1340 1.000 2.8 0 PB1 4.230 114 10 2.153 129.4 101 8 2.366 126.7 68.3 66 5 2.385 101.2 128.6 90.1 0 7 2.391 24.3 152.3 116.9 79.0 56 4 2.430 92.5 97.2 139.1 69.0 93.9 52 3 2.511 75.9 61.6 74.7 157.1 92.6 133.3 29 1 2.528 146.1 24.4 73.5 106.2 168.8 79.2 86.0 0 5 2.536 24.7 152.4 129.3 76.7 13.1 80.9 99.7 157.2 110 9 2.550 81.0 104.9 46.8 89.4 70.9 155.9 67.8 118.5 83.9 85 7 2.579 97.5 128.9 101.8 12.7 78.1 56.7 167.4 104.7 72.9 100.9 76 6 2.628 152.0 66.5 30.0 72.1 132.5 109.3 99.6 58.6 140.2 71.9 0 4 2.832 49.6 84.5 134.7 134.5 73.3 77.5 60.4 96.5 68.2 113.8 39 2 2.968 145.9 74.1 82.3 56.6 132.4 56.9 135.0 50.1 124.5 120.1 21 CR 3.520 176.3 47.0 54.6 82.1 159.4 87.1 101.7 30.3 158.2 100.8 25 CR 3.662 60.6 69.1 103.3 161.5 83.4 106.6 28.9 90.1 84.9 90.5 55 4 3.836 59.5 153.4 86.6 56.8 36.4 107.8 104.5 154.3 45.2 49.1 12 PB2 3.862 61.5 165.4 114.6 39.7 40.8 71.0 132.8 141.0 36.9 85.6 44 2 3.914 123.2 50.7 22.5 111.9 124.4 141.6 55.8 63.4 137.4 55.6 26 CR 3.966 61.2 127.2 69.7 81.1 47.3 135.6 77.7 142.4 60.1 24.0 CR 0.94 0.2946 0.8675 0.2046 1.000 2.5 20 6 1.839 7 1 1.847 86.4 12 3 1.902 174.0 87.8 16 4 1.936 92.5 96.4 89.5 29 9 1.937 95.7 166.4 89.6 96.9 10 2 2.040 79.7 74.6 97.4 168.4 92.5 32 10 2.588 71.3 19.8 103.3 84.3 167.1 85.0 28 8 2.887 19.9 72.8 156.6 80.0 112.4 90.1 55.1 25 7 2.930 52.7 122.0 132.4 54.1 68.8 124.3 102.2 55.3 19 5 3.483 53.5 119.9 131.4 51.2 71.3 126.8 100.1 54.7 3.2 4 PB2 3.520 46.8 43.6 127.3 111.1 132.0 57.4 37.5 41.9 96.6 96.4 11 2 3.523 49.6 76.8 130.1 46.8 114.7 122.6 57.4 33.6 45.9 43.4 33 10 3.525 145.6 123.7 40.2 69.9 59.2 121.1 132.4 146.6 94.1 93.9 6 PB2 3.662 142.3 94.3 39.7 49.9 92.2 136.7 98.3 127.0 97.3 95.3 2 PB1 3.730 122.3 48.1 52.2 121.4 121.1 57.9 68.0 116.7 170.1 167.5 27 8 3.806 86.5 90.6 92.2 172.8 76.2 17.1 102.0 100.8 120.6 123.6 26 8 3.848 104.0 165.9 81.5 92.6 9.1 97.6 174.2 119.6 72.0 74.2 1 PB1 3.897 130.1 136.7 54.6 103.1 36.1 88.5 156.0 148.2 100.5 102.3 17 5 3.972 44.5 75.2 132.2 135.9 97.0 35.3 73.8 56.0 93.4 95.1 13 3 3.987 130.9 96.1 48.5 135.4 72.4 54.4 115.6 144.5 141.1 143.7
1 1.36 0.4943 0.2593 -0.1809 1.000 2.0 0 PB1 2.851 0 PB2 4.754 61.7 0 10 1.056 133.2 78.2 22 CR 1.847 103.0 144.7 123.8 37 2 2.362 74.0 88.3 130.7 56.4 75 6 2.524 134.7 120.0 85.1 46.7 61.2 13 PB2 2.528 113.1 59.8 57.5 106.1 74.7 62.7 48 3 2.600 65.9 127.4 149.1 47.0 73.2 93.6 146.6 59 4 2.821 124.4 171.4 93.6 43.0 99.1 61.1 117.9 59.6 100 8 2.930 155.6 109.8 58.6 70.2 83.2 26.6 50.7 116.0 67.2 5 PB1 2.968 117.6 104.5 48.6 110.6 165.6 105.9 106.0 102.9 67.6 86.3 0 6 3.092 67.3 82.3 85.2 123.2 140.1 153.1 129.8 82.1 94.6 136.4 0 5 3.153 42.6 29.4 90.7 145.3 100.2 148.6 89.0 105.3 150.3 137.5 0 4 3.161 64.6 35.1 96.4 110.2 54.2 92.4 49.0 114.5 150.7 95.0 50 3 3.438 131.7 72.4 10.7 123.7 120.6 80.8 46.8 158.3 99.9 54.7 99 8 3.474 85.1 78.6 63.0 134.4 158.8 140.0 111.6 101.6 95.5 116.8 41 2 3.586 169.5 127.0 51.6 73.1 110.3 49.2 77.5 105.6 46.3 32.9 0 7 3.653 38.5 29.5 95.1 140.7 95.4 146.5 89.2 102.4 151.7 139.0 89 7 3.692 93.4 120.5 86.6 90.3 138.8 115.4 143.9 66.0 55.7 109.8 107 9 3.758 97.7 138.1 128.9 7.0 49.9 48.2 104.5 46.0 49.8 73.2 2 1.35 -0.2156 0.1972 -0.0096 1.000 4.0 0 PB1 2.446 0 PB2 4.946 58.7 0 8 1.950 52.2 74.4 23 CR 2.040 113.9 129.9 145.1 28 1 2.362 145.3 106.0 160.2 49.0 0 6 2.491 83.9 84.3 136.1 46.6 62.6 0 7 2.575 53.8 4.9 71.7 130.2 110.0 83.8 0 5 2.590 51.7 15.0 80.6 117.7 104.9 71.2 12.9 55 4 2.613 105.1 50.1 87.3 127.2 78.7 107.8 54.7 63.2 79 6 2.720 59.6 53.3 26.8 171.3 139.7 133.5 52.1 63.8 61.5 106 9 2.874 80.4 130.1 103.7 42.3 91.2 62.5 126.1 115.6 168.8 129.0 49 3 2.963 133.8 163.1 121.7 39.5 57.1 86.0 166.5 156.3 121.0 139.8 12 PB2 2.968 99.7 51.3 124.4 87.3 55.2 56.7 54.9 50.3 51.1 99.3 117 10 3.045 108.1 98.0 56.2 127.7 104.7 167.1 99.5 112.4 65.3 54.4 113 10 3.153 132.7 92.3 159.9 54.8 14.7 54.5 95.9 90.3 72.6 133.6 0 9 3.249 51.9 100.1 33.9 111.2 153.8 119.5 96.1 99.9 120.8 60.5 47 3 3.253 103.3 134.8 63.7 95.0 107.8 138.3 133.7 144.1 109.6 81.7 26 CR 3.523 86.8 52.6 55.0 157.5 108.9 133.5 54.8 67.5 32.7 31.0 99 8 3.539 90.0 75.3 140.3 54.7 55.3 15.4 76.0 64.4 92.6 128.1 33 1 3.586 103.1 82.7 54.7 139.7 105.5 158.9 84.5 97.4 51.3 44.6 3 1.30 0.5555 -0.0298 0.1542 1.000 3.2 0 PB1 3.197 20 CR 1.902 96.4 116 10 2.408 48.3 109.2 18 PB2 2.511 100.1 111.4 51.9 30 1 2.600 136.7 45.2 147.0 111.4 63 4 2.702 106.2 45.8 82.8 65.7 64.3 0 9 2.704 51.4 45.7 72.5 107.5 90.2 64.8 105 9 2.821 120.1 142.4 90.8 56.8 102.1 109.5 162.7 96 8 2.960 81.8 160.4 55.4 50.4 141.3 115.9 126.2 39.5 38 2 2.963 89.8 43.1 131.4 153.9 49.7 88.5 60.7 136.6 155.6 3 PB1 2.971 120.3 97.5 151.5 127.0 61.1 124.3 124.1 73.1 100.3 64.4 48 3 3.076 56.5 103.9 99.1 139.8 106.9 146.6 83.8 103.9 91.4 65.2 37 2 3.253 58.7 153.3 63.0 84.5 149.7 144.5 110.0 64.2 36.2 120.9 31 1 3.438 53.0 109.7 4.7 47.2 144.7 80.5 75.2 87.8 53.9 135.0 93 7 3.545 141.9 94.0 93.7 42.4 72.0 58.4 122.8 52.1 76.5 121.5 115 10 3.547 140.5 45.2 139.8 102.9 8.8 57.0 91.0 99.4 137.2 57.1 77 6 3.736 98.7 2.9 112.0 112.5 42.4 46.8 48.4 140.5 162.3 41.7 72 5 3.859 137.5 88.8 90.2 40.4 71.8 51.2 115.3 58.7 79.9 120.2 107 9 3.870 89.9 132.8 109.6 113.4 103.2 163.9 127.8 61.5 66.8 90.4 80 6 3.926 75.3 146.0 41.4 41.3 146.9 104.1 113.6 50.4 15.0 162.6 4 1.22 0.6248 0.3952 0.1002 1.000 2.2 0 PB1 3.222 0 PB2 2.832 88.4 25 CR 1.936 99.0 98.6 84 7 2.382 126.1 144.6 84.8 13 PB2 2.430 104.3 102.5 148.8 64.8 37 2 2.613 64.6 149.0 100.5 61.8 72.0 52 3 2.702 94.4 53.9 44.7 121.5 149.8 138.2 65 5 2.726 122.4 143.6 95.2 10.5 54.7 58.0 131.5 80 6 2.729 94.7 140.8 42.3 53.7 114.4 61.2 86.9 62.0 31 1 2.821 58.4 96.7 40.6 107.8 154.0 82.5 56.1 115.2 54.1 111 9 2.898 140.4 94.5 41.6 65.0 113.4 116.0 57.6 74.3 59.6 82.1 0 5 3.020 41.1 51.2 117.1 153.8 94.0 98.0 84.6 143.8 133.7 84.3 116 10 3.074 44.8 108.0 56.9 103.3 134.1 64.1 75.5 107.7 54.0 20.0 0 7 3.137 41.9 46.9 106.9 163.4 104.2 103.7 74.4 154.1 128.7 76.4 0 1 3.161 53.0 104.9 141.9 92.6 51.8 47.1 144.2 83.4 108.0 106.3 96 8 3.185 78.1 154.7 63.2 55.8 101.6 37.7 105.4 59.8 24.1 58.0 67 5 3.224 150.2 82.6 110.4 63.6 51.0 112.9 102.8 61.1 110.2 150.8 56 4 3.310 99.1 45.8 139.1 112.6 56.7 120.4 97.5 105.4 165.1 139.4 114 10 3.388 124.1 39.2 77.7 109.3 105.1 171.2 44.9 113.4 114.2 100.9 0 10 3.442 66.4 95.3 159.6 92.5 38.3 60.9 145.4 82.2 121.7 122.9 5 1.20 0.0933 0.3719 0.0113 1.000 3.1 0 PB1 2.199 0 PB2 2.536 126.4 0 7 0.581 90.0 69.1 12 PB2 2.385 129.7 103.3 103.1 0 2 2.590 60.8 149.6 82.1 73.1 55 4 2.726 108.8 93.5 48.6 56.3 58.8 79 6 2.809 60.5 95.8 31.1 113.1 60.4 59.1 0 6 2.957 78.1 150.3 133.6 57.8 52.9 93.0 112.3 0 8 2.976 41.8 121.1 56.5 109.0 40.3 67.8 26.1 88.1 0 4 3.020 74.4 60.5 96.2 148.3 135.1 143.8 96.4 122.2 102.6 85 7 3.040 175.7 54.2 94.1 50.6 121.2 75.1 123.8 100.1 142.3 103.7 66 5 3.054 173.0 49.4 83.1 53.9 119.3 67.5 113.0 107.8 133.1 104.8 0 1 3.153 61.3 112.9 146.6 108.4 96.4 152.7 121.4 60.7 101.7 61.5 56 4 3.224 131.9 48.1 116.5 84.9 154.3 118.9 143.6 104.1 165.2 63.9 0 10 3.338 79.7 104.0 160.7 96.1 106.4 150.5 139.8 60.4 119.5 65.3 99 8 3.361 99.2 127.6 142.8 44.7 71.6 94.6 131.9 22.9 109.8 121.0 110 9 3.399 108.1 48.2 22.0 99.5 101.8 55.2 52.6 148.1 78.4 89.1 108 9 3.473 124.7 94.6 143.3 47.2 103.3 102.9 159.6 55.8 142.9 103.9 26 CR 3.483 91.8 80.8 16.2 89.6 69.1 33.6 31.8 118.4 52.4 112.2 28 1 3.928 92.6 137.2 97.4 38.2 35.5 53.1 88.6 40.0 74.5 161.1 6 1.11 -0.0615 0.2447 -0.2463 1.000 3.0 0 PB1 3.301 99 8 1.315 132.5 23 CR 1.839 89.6 131.8 89 7 2.331 115.7 91.2 88.4 0 2 2.491 47.5 134.5 53.7 132.5 28 1 2.524 103.1 94.2 46.9 120.3 56.2 5 PB1 2.576 115.2 49.2 141.5 54.9 161.8 139.0 12 PB2 2.628 88.1 63.9 102.6 154.1 70.8 58.7 107.0 113 10 2.652 118.9 70.9 67.6 119.2 75.6 23.4 117.1 48.1 43 2 2.720 170.1 41.9 99.4 60.7 142.2 86.2 54.9 94.1 69.0 60 4 2.729 129.6 97.8 45.1 55.5 98.4 64.8 98.4 117.8 69.7 57.3 106 9 2.801 68.3 156.6 43.5 67.2 65.5 89.6 117.0 135.6 111.1 115.7 71 5 2.809 119.5 84.1 94.8 7.4 139.9 122.9 48.0 147.6 118.3 55.8 0 5 2.957 40.7 96.1 109.6 147.1 56.0 91.2 108.2 50.1 93.5 137.3 108 9 3.040 105.7 26.8 149.3 107.6 119.5 102.9 54.3 52.9 81.7 68.4 0 1 3.092 52.8 95.5 132.4 84.6 100.1 153.1 62.4 103.9 151.9 117.3 0 10 3.182 68.6 76.2 151.5 85.1 113.9 153.4 47.9 95.1 138.7 101.5 0 7 3.384 39.8 100.1 102.7 151.6 49.2 85.1 115.3 48.8 89.3 140.1 49 3 3.736 89.8 130.2 3.0 91.0 52.3 44.0 143.5 99.6 64.9 99.3 51 3 3.926 127.0 35.8 104.0 115.7 101.9 59.8 84.8 39.1 36.9 55.1 7 1.09 0.0539 0.3749 0.0671 1.000 3.3 0 PB1 2.274 0 PB2 2.391 130.1 0 5 0.581 75.2 97.8 79 6 2.331 68.0 114.5 141.4 55 4 2.382 119.2 107.0 120.8 70.7 0 2 2.575 60.2 169.7 85.0 67.1 63.5 12 PB2 2.579 117.4 101.9 64.3 124.1 58.5 70.3 0 8 2.699 46.0 141.3 113.1 29.1 77.4 43.3 112.0 110 9 2.868 125.8 57.1 153.6 64.2 66.2 118.3 109.8 93.1 26 CR 2.930 106.0 95.8 160.6 38.9 41.1 79.3 99.4 61.6 39.0 66 5 3.040 161.1 50.4 85.9 130.7 71.8 120.3 52.9 149.0 71.9 92.2 85 7 3.135 150.5 53.6 75.3 141.0 78.0 118.3 48.3 154.7 82.3 102.2 0 4 3.137 71.1 59.8 73.2 104.4 163.4 130.3 130.8 106.4 97.3 125.9 0 6 3.384 68.1 133.0 39.2 112.5 89.6 47.0 50.1 84.6 155.6 122.5 56 4 3.522 116.4 43.5 55.0 156.2 119.5 136.4 76.2 162.3 98.8 134.3 52 3 3.545 94.6 45.0 118.7 76.4 116.7 141.2 146.1 97.9 50.9 80.6 0 1 3.653 51.3 101.7 28.3 119.1 141.6 85.4 91.2 95.8 152.1 157.2 31 1 3.692 53.5 85.1 109.1 56.5 125.5 103.4 170.8 63.6 79.0 85.7 99 8 3.840 85.3 114.7 31.9 130.5 89.3 63.4 37.1 103.7 146.7 128.8 108 9 3.954 105.4 85.6 31.6 158.7 97.3 91.8 39.3 132.7 128.2 137.0 8 1.03 -0.0694 0.1605 0.1729 1.000 4.2 0 PB1 1.984 0 PB2 4.804 61.4 79 6 1.315 100.8 44.3 0 2 1.950 76.9 82.6 111.4 0 9 1.959 82.3 137.0 135.6 112.4 16 PB2 2.366 148.0 130.2 86.1 129.9 71.5 117 10 2.544 159.9 110.0 79.9 84.2 111.4 51.8 0 7 2.699 55.6 18.1 59.7 64.9 137.8 145.1 110.1 26 CR 2.887 116.8 55.6 28.4 91.4 153.0 83.5 56.5 63.2 33 1 2.930 152.4 92.3 59.2 92.4 125.2 55.8 20.7 96.8 37.0 47 3 2.960 129.8 155.5 129.2 80.1 66.4 54.9 51.2 142.8 107.7 71.4 0 5 2.976 47.6 26.9 69.9 59.1 129.8 155.4 116.1 10.3 72.9 105.0 116 10 3.140 50.0 70.5 79.8 126.7 68.5 101.9 147.7 80.3 108.0 132.4 55 4 3.185 99.3 52.8 58.1 55.0 165.9 110.5 63.8 46.9 36.8 54.8 70 5 3.361 111.5 95.1 61.0 169.0 76.5 45.2 86.5 113.2 78.5 76.9 31 1 3.474 58.5 58.8 62.4 130.5 83.6 99.5 135.0 72.2 90.6 116.8 40 2 3.539 127.7 74.4 30.2 125.4 118.2 56.2 59.8 89.0 35.2 41.5 23 CR 3.806 73.7 96.3 129.1 17.9 94.6 125.4 90.2 78.2 108.6 103.2 91 7 3.840 116.7 96.8 60.2 164.3 78.7 41.1 81.3 114.9 75.6 71.9 20 CR 3.848 76.5 128.4 129.3 116.9 9.0 75.1 118.8 131.1 151.4 130.4 9 1.03 0.0674 -0.0010 0.1816 1.000 4.3 0 PB1 2.595 20 CR 1.937 117.9 0 8 1.959 49.3 162.0 16 PB2 2.550 108.8 123.6 61.7 0 3 2.704 74.2 44.7 118.8 139.5 77 6 2.801 155.3 40.8 154.7 95.7 85.4 47 3 2.821 112.2 123.7 74.1 55.5 162.7 83.8 95 7 2.868 150.6 72.2 112.9 52.0 105.6 48.5 77.1 38 2 2.874 105.3 45.2 142.6 143.1 64.1 52.0 98.6 100.5 63 4 2.898 118.6 41.5 128.1 89.3 57.5 57.2 125.4 48.8 86.5 116 10 3.032 49.9 87.5 74.5 100.5 49.2 123.9 147.2 107.0 112.2 69.6 79 6 3.040 53.7 147.6 17.6 55.2 111.6 150.2 84.0 102.1 157.3 110.5 0 2 3.249 47.9 151.7 33.7 84.0 119.1 141.6 64.4 133.5 109.7 160.5 74 5 3.399 147.0 76.1 108.6 47.9 107.9 52.8 76.0 4.4 104.8 50.5 1 PB1 3.449 109.9 83.7 111.7 109.0 107.4 62.7 55.5 98.3 44.4 118.5 70 5 3.473 94.3 101.5 70.2 43.4 96.7 101.9 98.8 56.3 146.4 60.0 106 9 3.539 66.4 103.6 83.2 123.6 95.2 102.7 74.0 141.3 59.8 144.7 117 10 3.735 87.7 153.9 39.4 33.9 156.2 115.4 40.1 82.7 137.4 123.0 30 1 3.758 114.9 6.6 162.6 129.5 43.8 42.2 122.8 77.6 38.9 48.1 97 8 3.836 87.9 74.5 113.6 138.8 80.6 75.0 83.4 121.4 29.6 116.0 10 0.89 0.4903 0.3356 -0.2457 1.000 1.7 0 PB1 3.655 0 PB2 4.654 59.8 0 1 1.056 34.7 89.0 13 PB2 2.153 97.9 62.0 98.1 5 PB1 2.404 108.7 119.6 112.2 149.6 50 3 2.408 141.8 82.5 164.6 66.5 83.2 100 8 2.544 130.5 121.8 100.7 59.8 109.2 73.4 22 CR 2.588 71.0 119.9 36.4 95.5 106.8 141.7 68.5 75 6 2.652 102.6 119.9 71.5 65.4 120.4 101.7 29.2 41.1 108 9 3.032 97.9 67.2 129.5 107.2 55.6 58.3 129.8 156.1 159.0 41 2 3.045 146.0 152.0 112.6 96.2 69.8 72.2 39.6 77.0 56.5 107.2 59 4 3.074 96.0 155.2 66.3 121.7 70.9 122.1 68.3 38.8 56.3 126.4 99 8 3.140 78.1 83.3 99.5 140.6 39.2 92.3 147.7 119.1 154.0 37.0 37 2 3.153 54.7 82.0 34.6 64.9 143.8 130.7 76.0 40.1 49.9 147.1 0 6 3.182 57.2 83.1 75.5 144.8 52.7 115.9 155.0 98.4 138.7 58.5 0 5 3.338 36.3 31.9 70.8 91.1 101.5 106.7 148.9 107.2 131.5 65.9 56 4 3.388 89.7 30.3 118.8 56.3 108.2 52.3 107.2 144.0 121.5 53.3 0 4 3.442 53.9 37.3 65.9 44.5 154.2 100.2 96.2 86.4 84.2 104.4 24 CR 3.525 113.8 60.0 147.8 76.1 79.7 30.6 103.2 170.6 129.8 33.3 48 3 3.547 48.7 107.9 22.1 116.9 92.2 169.5 99.5 31.4 72.5 126.2
Examination of the connectivity list suggest that peaks Q1, Q2,
Q3, Q4, Q6, Q9 form a reasonable CrF6 octahedron with the Cr atom. Peak
number 10 is too close to peak 1, and should be eliminated. The same for
peak 8, too close to peak 6. Peaks 5 and 7 are 0.5 angstrom apart, but
one of them would make a good 7th fluorine atom, connected only to Pb atoms.
Possibly, a more clean Fourier difference synthesis would be obtained from a dataset reduced by using the overlap program.
We may have a look at the structure model by using the STRUPLO program,
reading the .res file generated by SHELXL-97, and reorganized :
TITL "PbF2/CrF3/HFaq" ??? P21/c CELL 1.54056 5.46255 11.20846 9.57362 90.0 91.19608 90.0 ZERR 2 0.001 0.001 0.001 0. 0.001 0. LATT 1 SYMM X,0.5-Y,0.5+Z SFAC PB CR F UNIT 8 4 28 L.S. 4 ACTA BOND FMAP 2 PLAN -10 WGHT 0.100000 FVAR 0.49932 PB1 1 0.21949 0.18912 0.05528 11.00000 0.01534 PB2 1 0.23830 0.56038 0.13397 11.00000 0.01790 CR 2 0.29464 0.86749 0.20458 11.00000 0.00546 Q1 1 0.4943 0.2593 -0.1809 11.00000 0.05 1.36 Q2 1 -0.2156 0.1972 -0.0096 11.00000 0.05 1.35 Q3 1 0.5555 -0.0298 0.1542 11.00000 0.05 1.30 Q4 1 0.6248 0.3952 0.1002 11.00000 0.05 1.22 Q5 1 0.0933 0.3719 0.0113 11.00000 0.05 1.20 Q6 1 -0.0615 0.2447 -0.2463 11.00000 0.05 1.11 Q9 1 0.0674 -0.0010 0.1816 11.00000 0.05 1.03 HKLF 3 ENDThe Struplo output is very conclusive, since the CrF6 octahedra appear with the default option :
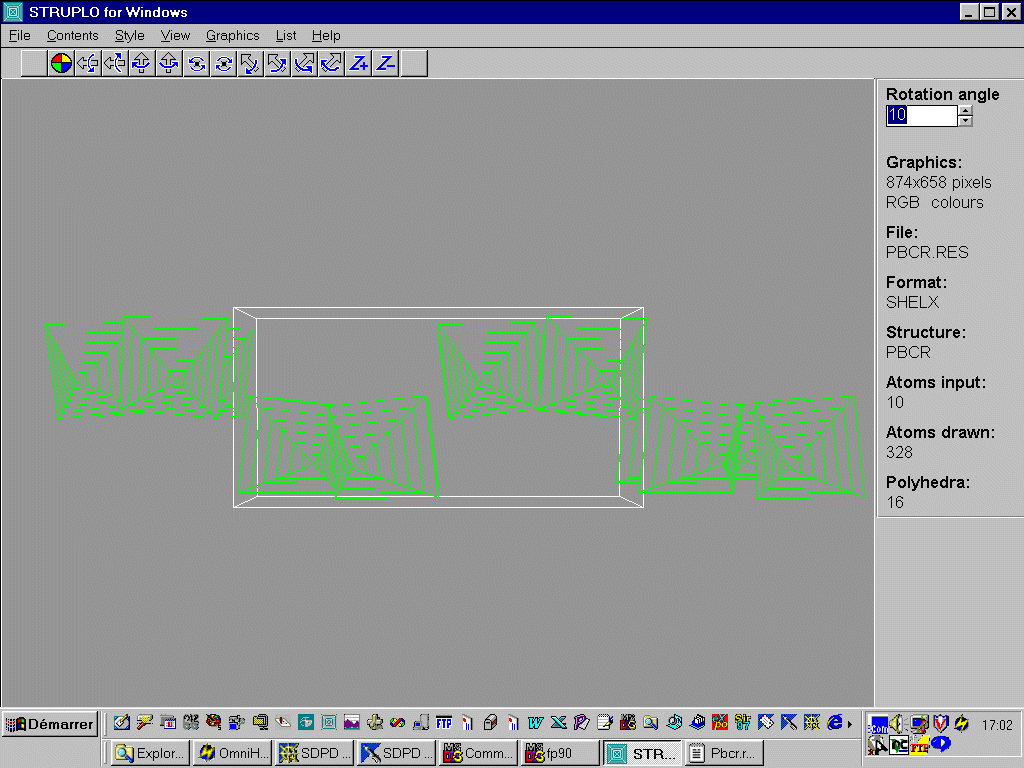
You may modify the Struplo file, once saved, for making the Pb and "isolated" fluorine atoms to appear.
You will probably encounter cases where it will be better to enter only one supplementary atom in the Rietveld refinement, and to perform a new Fourier difference synthesis, etc. Here we will add all these suggested fluorine atoms in one run, and see what happens :
The new .pcr file becomes :
COMM PbCr ?? ! Files => DAT-file: pbcr, PCR-file: pbcr !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 2 11 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.40 0.40 0.40 0.80 5.0000 0.0100 150.0000 0.000 0.000 ! ! 2Theta/TOF Background 5.00 530.76 6.92 489.66 9.08 446.92 13.88 389.39 16.76 358.16 22.04 333.50 27.57 312.13 39.81 287.47 84.94 256.24 112.07 246.38 150.00 267.75 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 49 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0531 121.00 0.0000 0.00 0.0000 0.00 0.000000 0.00 0 ! ! Data for PHASE number: 1 ==> Current R_Bragg: 12.93 PbCr ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 10 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/C <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes PB1 PB+2 0.21935 0.18913 0.05528 1.02945 1.00000 0 0 0 161.00 171.00 181.00 191.00 0.00 PB2 PB+2 0.23843 0.56035 0.13402 1.23951 1.00000 0 0 0 201.00 211.00 221.00 231.00 0.00 CR CR+3 0.29400 0.86764 0.20427 0.31564 1.00000 0 0 0 241.00 251.00 261.00 271.00 0.00 Q1 F-1 0.4943 0.2593 -0.1809 1.20000 1.00000 0 0 0 281.00 291.00 301.00 491.00 0.00 Q2 F-1 -0.2156 0.1972 -0.0096 1.20000 1.00000 0 0 0 311.00 321.00 331.00 491.00 0.00 Q3 F-1 0.5555 -0.0298 0.1542 1.20000 1.00000 0 0 0 341.00 351.00 361.00 491.00 0.00 Q4 F-1 0.6248 0.3952 0.1002 1.20000 1.00000 0 0 0 371.00 381.00 391.00 491.00 0.00 Q5 F-1 0.0933 0.3719 0.0113 1.20000 1.00000 0 0 0 401.00 411.00 421.00 491.00 0.00 Q6 F-1 -0.0615 0.2447 -0.2463 1.20000 1.00000 0 0 0 431.00 441.00 451.00 491.00 0.00 Q9 F-1 0.0674 -0.0010 0.1816 1.20000 1.00000 0 0 0 461.00 471.00 481.00 491.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.27166E-03 0.6467 0.0000 0.0000 0.0000 0.0000 0 11.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.02743 -0.01386 0.00608 0.00000 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 0.00 0.00 0.00 0.00 ! a b c alpha beta gamma 5.463544 11.211061 9.575928 90.000000 91.194016 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.15855-0.01270 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00 ! ! Data for PHASE number: 2 ==> Current R_Bragg: 4.52 CrF3 ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 2 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! R -3 C <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR CR+3 0.00000 0.00000 0.00000 0.50000 0.06000 0 0 0 0.00 0.00 0.00 0.00 0.00 F F-1 0.60000 0.00000 0.25000 0.80000 0.18000 0 0 0 0.00 0.00 0.00 0.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.15102E-01 0.6467 0.0000 0.0000 0.0000 0.0000 0 151.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.02743 -0.01386 0.00608 0.00000 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 0.00 0.00 0.00 0.00 ! a b c alpha beta gamma 4.977706 4.977706 13.147063 90.000000 90.000000 120.000000 131.00000 131.00000 141.00000 0.00000 0.00000 131.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.15855-0.01270 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00The refinement starts with quite better R values :
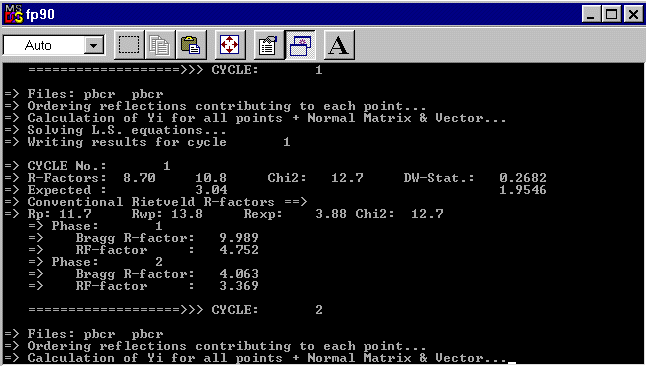
And quickly, the reliabilities decrease :
******************************************************* *** PROGRAM FULLPROF (Version 3.3 - Aug97-LLB JRC) *** ******************************************************* Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 03/13/00 Time: 16:52:16.61 => PCR file code: pbcr => DAT file code: pbcr => Title: PbCr ?? ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Base of peaks: 2.0*HW* 15.00 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 150.0000 Step: 0.0100 No. of points: 14501 => Crystal Structure Refinement for phase: 1 => Crystal Structure Refinement for phase: 2 => Scor: 3.5580 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 49 ------------------------------------------------------------------------------ => Phase No. 1 PbCr ?? P 21/C ------------------------------------------------------------------------------ => No. of reflections: 2405 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. PB1 0.21898( 11) 0.18752( 6) 0.05506( 6) 1.390( 12) 1.000( 0) PB2 0.23967( 11) 0.56103( 6) 0.13503( 6) 1.469( 12) 1.000( 0) CR 0.29952( 41) 0.86850( 24) 0.20624( 26) 0.790( 50) 1.000( 0) Q1 0.46696( 118) 0.25832( 60) -0.21118( 72) 1.364( 62) 1.000( 0) Q2 -0.21072( 110) 0.17989( 59) -0.02003( 63) 1.364( 62) 1.000( 0) Q3 0.55251( 114) -0.03279( 67) 0.14646( 62) 1.364( 62) 1.000( 0) Q4 0.60571( 111) 0.40343( 59) 0.10639( 64) 1.364( 62) 1.000( 0) Q5 0.12437( 111) 0.38388( 61) 0.00251( 70) 1.364( 62) 1.000( 0) Q6 -0.04613( 121) 0.24238( 62) -0.25853( 69) 1.364( 62) 1.000( 0) Q9 0.07832( 122) -0.00044( 59) 0.20126( 69) 1.364( 62) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 5.46269 0.00002 11.20868 0.00004 9.57393 0.00004 90.00000 0.00000 91.19644 0.00024 90.00000 0.00000 => Overall scale factor : 0.000309702 0.000000421 => ETA(p-V) or M(P-VII) : 0.77432 0.00305 => Overall Tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.01869 0.00030 -0.00475 0.00024 0.00274 0.00005 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.05474 0.00260 0.00007 0.00039 0.00000 0.00000 0.00000 0.00000 => X and Y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ------------------------------------------------------------------------------ => Phase No. 2 CrF3 R -3 C ------------------------------------------------------------------------------ => No. of reflections: 140 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.500( 0) 0.060( 0) F 0.60000( 0) 0.00000( 0) 0.25000( 0) 0.800( 0) 0.180( 0) ==> PROFILE PARAMETERS: => Cell parameters : 4.97683 0.00002 4.97683 0.00002 13.14515 0.00009 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => Overall scale factor : 0.014576116 0.000034674 => ETA(p-V) or M(P-VII) : 0.77432 0.00305 => Overall Tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.01869 0.00030 -0.00475 0.00024 0.00274 0.00005 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.05474 0.00260 0.00007 0.00039 0.00000 0.00000 0.00000 0.00000 => X and Y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0364 0.0003 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13950 => Rp: 4.66 Rwp: 6.32 Rexp: 3.04 Chi2: 4.31 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 6.27 Rwp: 8.05 Rexp: 3.88 Chi2: 4.31 => Deviance: 0.592E+05 Dev* : 4.241 => DW-Stat.: 0.6093 DW-exp: 1.9546 => N-sigma of the GoF: 276.467 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13375 => Rp: 4.66 Rwp: 6.33 Rexp: 3.00 Chi2: 4.44 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 6.19 Rwp: 8.00 Rexp: 3.79 Chi2: 4.44 => Deviance: 0.585E+05 Dev* : 4.369 => DW-Stat.: 0.6164 DW-exp: 1.9538 => N-sigma of the GoF: 281.582 => Phase: 1 => Bragg R-factor: 3.08 Vol: 586.080 Fract(%): 0.00 => Rf-factor= 2.97 ATZ: 0.000 Brindley: 1.0000 => Phase: 2 => Bragg R-factor: 2.55 Vol: 281.969 Fract(%): 0.00 => Rf-factor= 2.27 ATZ: 0.000 Brindley: 1.0000We may try now to improve the fit by using the parametrized polynomial representation of the background, like in the previous Na5Al3F14 case. Of course, performing a final Fourier difference synthesis is necessary, but you will not find any new meaningful peak.
After several runs, adding more and more parameters, including separated
U, V, W values for the 2 phases, here is the final .sum FULLPROF file :
******************************************************** ** PROGRAM FULLPROF.98 (Version 0.2 - Mar98-LLB JRC) ** ******************************************************** Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 14/03/2000 Time: 08:51:07.770 => PCR file code: pbcr => DAT file code: pbcr => Title: PbCr ?? ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 => Base of peaks: 2.0*HW* 15.00 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 150.0000 Step: 0.0100 No. of points: 14501 => Crystal Structure Refinement for phase: 1 => Crystal Structure Refinement for phase: 2 => Scor: 1.5596 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 65 ------------------------------------------------------------------------------ => Phase No. 1 PbCr ?? P 21/C ------------------------------------------------------------------------------ => No. of reflections: 2403 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. PB1 0.21910( 6) 0.18751( 3) 0.05496( 3) 0.881( 7) 1.000( 0) PB2 0.23967( 6) 0.56094( 3) 0.13525( 3) 0.846( 8) 1.000( 0) CR 0.30080( 22) 0.86734( 12) 0.20739( 14) 0.430( 26) 1.000( 0) Q1 0.46853( 65) 0.26204( 34) -0.21435( 40) 1.198( 34) 1.000( 0) Q2 -0.21051( 61) 0.17795( 33) -0.02293( 35) 1.198( 34) 1.000( 0) Q3 0.55267( 63) -0.03313( 37) 0.14533( 34) 1.198( 34) 1.000( 0) Q4 0.60397( 61) 0.40591( 33) 0.10458( 35) 1.198( 34) 1.000( 0) Q5 0.12525( 62) 0.38461( 33) 0.00372( 39) 1.198( 34) 1.000( 0) Q6 -0.04344( 67) 0.24117( 34) -0.26094( 38) 1.198( 34) 1.000( 0) Q9 0.08395( 68) -0.00202( 33) 0.19897( 38) 1.198( 34) 1.000( 0) ==> PROFILE PARAMETERS: => Cell parameters : 5.46257 0.00001 11.20845 0.00002 9.57377 0.00002 90.00000 0.00000 91.19705 0.00013 90.00000 0.00000 => overall scale factor : 0.000296038 0.000000263 => Eta(p-v) or m(p-vii) : 0.35651 0.00310 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.00375 0.00007 0.00121 0.00008 0.00247 0.00002 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.04592 0.00151 0.00260 0.00025 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.01247 0.00006 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ------------------------------------------------------------------------------ => Phase No. 2 CrF3 R -3 C ------------------------------------------------------------------------------ => No. of reflections: 140 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. CR 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.519( 10) 0.060( 0) F 0.59541( 14) 0.00000( 0) 0.25000( 0) 0.922( 18) 0.180( 0) ==> PROFILE PARAMETERS: => Cell parameters : 4.97679 0.00001 4.97679 0.00001 13.14501 0.00004 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => overall scale factor : 0.014619797 0.000029180 => Eta(p-v) or m(p-vii) : 0.88953 0.00446 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.01229 0.00025 -0.00377 0.00022 0.00225 0.00004 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.03297 0.00220 0.00042 0.00050 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00000 0.00000 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: 0.0352 0.0001 => Background Polynomial Parameters ==> 182.741 0.736442 -168.356 2.24982 469.410 6.25947 659.487 13.2592 -977.614 14.8567 -1453.49 21.8424 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 10 => MaxCycle: 10 => N-P+C: 13934 => Rp: 2.44 Rwp: 3.46 Rexp: 3.04 Chi2: 1.29 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 3.12 Rwp: 4.24 Rexp: 3.73 Chi2: 1.29 => Deviance: 0.181E+05 Dev* : 1.298 => DW-Stat.: 1.6189 DW-exp: 1.9570 => N-sigma of the GoF: 24.341 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 13402 => Rp: 2.42 Rwp: 3.42 Rexp: 3.00 Chi2: 1.30 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 3.05 Rwp: 4.17 Rexp: 3.66 Chi2: 1.30 => Deviance: 0.175E+05 Dev* : 1.305 => DW-Stat.: 1.6732 DW-exp: 1.9563 => N-sigma of the GoF: 24.505 => Phase: 1 => Bragg R-factor: 0.570 Vol: 586.045( 0.002) Fract(%): 0.00( 0.00) => Rf-factor=0.416 ATZ: 0.000 Brindley: 1.0000 => Phase: 2 => Bragg R-factor: 0.510 Vol: 281.962( 0.001) Fract(%): 0.00( 0.00) => Rf-factor=0.389 ATZ: 0.000 Brindley: 1.0000 => Run finished at: Date: 14/03/2000 Time: 09:10:28.340
Again, this is too much good !-).
GFOUR needs a .inp and a .fou files which are generated by FULLPROF
(FP98) by selecting the option FOU=4 at line 3 in the .pcr file. We start
at the step where Pb and Cr atoms aare already located and their coordinates
refined :
COMM PbCr ?? ! Files => DAT-file: pbcr, PCR-file: pbcrg !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 2 11 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 1 4 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 15.0000 0.7995 0.0000 80.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 10 0.01 0.60 0.60 0.60 0.80 5.0000 0.0100 150.0000 0.000 0.000 ! ! 2Theta/TOF Background 5.000 530.760 6.920 489.660 9.080 446.920 13.880 389.390 16.760 358.160 22.040 333.500 27.570 312.130 39.810 287.470 84.940 256.240 112.070 246.380 150.000 267.750 ! ! Excluded regions (LowT HighT) 1.00 10.00 150.00 170.00 ! ! 27 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE 0.0531 121.00 0.0000 0.00 0.0000 0.00 0.000000 0.00 0 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 12.93 !------------------------------------------------------------------------------- PbCr ?? ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 3 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! P 21/C <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes PB1 PB+2 0.21935 0.18913 0.05528 1.02946 1.00000 0 0 0 161.00 171.00 181.00 191.00 0.00 PB2 PB+2 0.23843 0.56035 0.13402 1.23951 1.00000 0 0 0 201.00 211.00 221.00 231.00 0.00 CR CR+3 0.29400 0.86764 0.20427 0.31564 1.00000 0 0 0 241.00 251.00 261.00 271.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.27166E-03 0.6467 0.0000 0.0000 0.0000 0.0000 0 11.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.02743 -0.01386 0.00608 0.00000 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 0.00 0.00 0.00 0.00 ! a b c alpha beta gamma 5.463544 11.211061 9.575928 90.000000 91.194016 90.000000 81.00000 91.00000 101.00000 0.00000 111.00000 0.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.15855-0.01270 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00 !------------------------------------------------------------------------------- ! Data for PHASE number: 2 ==> Current R_Bragg: 4.52 !------------------------------------------------------------------------------- CrF3 ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 2 0 0 1.0 0.0 0.0 0 0 0 0 0 0.00 0 5 0 ! R -3 C <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes CR CR+3 0.00000 0.00000 0.00000 0.50000 0.06000 0 0 0 0.00 0.00 0.00 0.00 0.00 F F-1 0.60000 0.00000 0.25000 0.80000 0.18000 0 0 0 0.00 0.00 0.00 0.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.15102E-01 0.6467 0.0000 0.0000 0.0000 0.0000 0 151.00000 21.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.02743 -0.01386 0.00608 0.00000 0.00000 0.00000 0.00000 0 51.00 41.00 31.00 0.00 0.00 0.00 0.00 ! a b c alpha beta gamma 4.977706 4.977706 13.147063 90.000000 90.000000 120.000000 131.00000 131.00000 141.00000 0.00000 0.00000 131.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.15855-0.01270 0.00000 0.00000 0.00 0.00 61.00 71.00 0.00 0.00
The pbcr.inp file which is now built as a default file (in which
you will have to change some options) is the following :
! Template file to be used with the program FOURIER ! Title of the job Titl PbCr ?? ! Cell parameters Cell 5.4635 11.2111 9.5759 90.0000 91.1940 90.0000 ! Atoms in the asymmetric unit Atom PB1 0.21935 0.18913 0.05528 Atom PB2 0.23843 0.56035 0.13402 Atom CR 0.29400 0.86764 0.20427 ! Space group symbol Spgr P 21/C ! Type of Fourier synthesis Fourier Fobs -> Change Fobs by DIFF for a Fourier difference synthesis ! Name of the Fourier-reflection file File fou pbcrg.fou ! Format of the Fourier-reflection file Form h k l fo fc phase ! Standard output binary file File bin ! Peak search Scan 15 ! End of file EndAnd the .fou file contains the observed and calculated structure factors, as well as the phases :
Phase no.: 1 PbCr ?? 0 1 1 63.1651 62.9904 180.0000 0 2 0 9.8212 10.4816 180.0000 1 0 0 50.9617 51.6332 0.0000 1 1 0 70.4325 70.6533 180.0000 0 2 1 106.0051 106.1186 180.0000 0 0 2 90.0828 89.8764 0.0000 1 1 -1 93.4067 93.9811 180.0000 0 1 2 25.4863 25.6405 180.0000 1 1 1 89.3754 89.5878 0.0000 1 2 0 33.4734 34.5056 180.0000 1 2 -1 197.7849 197.5818 180.0000 0 2 2 154.7034 154.5342 180.0000 1 0 -2 536.0651 535.5217 0.0000 etc
We may now start GFOUR, click on "File) and open the pbcrg.inp file :
Then, clicking on "Calculations", enad "Fourier", and a section of the Fourier map appears, with a lot of possibilities for visiting it in full details :
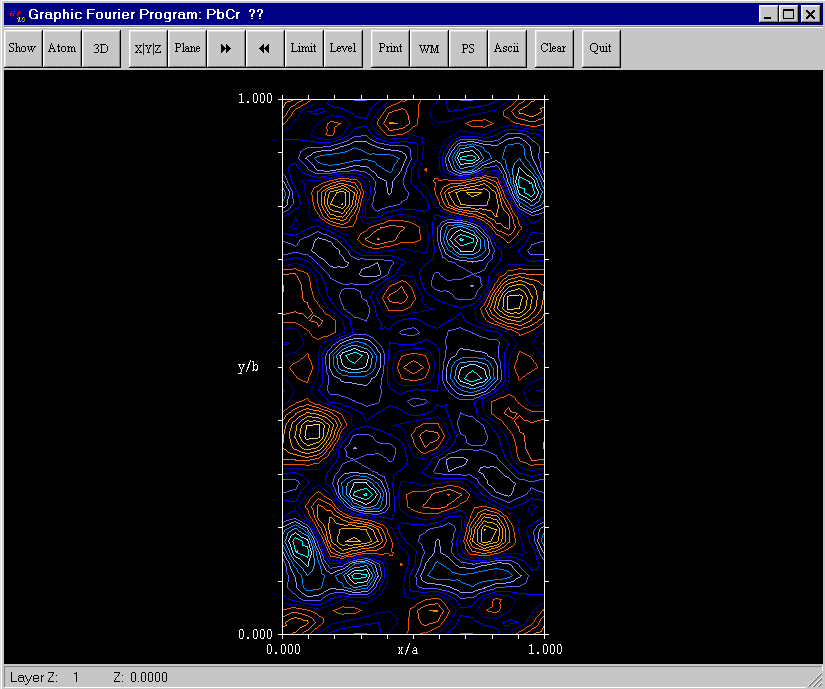
The summary of the calculation is in the pbcrg.out file, where we will
find our 15 peaks asked to be found by automatic search :
!***************************************************************************!
!**** FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ****!
!**** Copyright(c) 1999. Release: 01.06 ****!
!**** Javier Gonzalez-Platas & Juan Rodriguez-Carvajal ****!
!**** E:\sdpd-co Date: 14/03/2000 Time: 09:47:14 ****!
!***************************************************************************!
!---------------------------------------------------------------------------!
!---- FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ----!
!---- Copyright(c) 1999. Release: 01.06 ----!
!---- ----!
!---- Files Information ----!
!---------------------------------------------------------------------------!
Input Data: E:\sdpd-course-solutions\week-9\data\pbcrg.inp
Output Data: E:\sdpd-course-solutions\week-9\data\pbcrg.out
HKL- F Data: pbcrg.fou
Binary Data: E:\sdpd-course-solutions\week-9\data\pbcrg.bin
Peaks Data: E:\sdpd-course-solutions\week-9\data\pbcrg.pks
!---------------------------------------------------------------------------!
!---- FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ----!
!---- Copyright(c) 1999. Release: 01.06 ----!
!---- ----!
!---- Cell Parameters Information ----!
!---------------------------------------------------------------------------!
=> Direct cell parameters:
a = 5.4635 b = 11.2111 c = 9.5759
alpha = 90.000 beta = 91.194 gamma = 90.000
Direct Cell Volume = 586.4142
=> Reciprocal cell parameters:
a*= 0.183073 b*= 0.089197 c*= 0.104451
alpha*= 90.000 beta*= 88.806 gamma*= 90.000
Reciprocal Cell Volume = 0.00170528
=> Direct and Reciprocal Metric Tensors:
GD GR
29.8498 0.0000 -1.0902 0.033516 0.000000 0.000398
0.0000 125.6888 0.0000 0.000000 0.007956 0.000000
-1.0902 0.0000 91.6979 0.000398 0.000000 0.010910
=> Crystal_to_Orthonormal_Matrix Orthonormal_to_Crystal Matrix
Cr_Orth_cel Orth_Cr_cel
5.4623 0.0000 0.0000 0.183073 0.000000 0.000000
0.0000 11.2111 0.0000 0.000000 0.089197 0.000000
-0.1138 0.0000 9.5759 0.002177 0.000000 0.104429
!---------------------------------------------------------------------------!
!---- FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ----!
!---- Copyright(c) 1999. Release: 01.06 ----!
!---- ----!
!---- Space Group Information ----!
!---------------------------------------------------------------------------!
=> Space group: P 21/C
=> N. Space group: 14
=> Crystal System: Monoclinic
=> Laue Class: 2/m
=> Point Group: 2/m
=> Centrosymmetry: Centric
=> Bravais Lattice: P: { 000 }
=> Lattice Symbol: mP
=> Asymmetric unit: 0.000 <= x <= 1.000
0.000 <= y <= 0.250
0.000 <= z <= 1.000
=> Number of S.O.: 2
=> General multiplicity: 4
=> List of S.O. without inversion and lattice centring translations
=> SYMM( 1): x,y,z => SYMM( 2): -x,y+1/2,-z+1/2
!---------------------------------------------------------------------------!
!---- FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ----!
!---- Copyright(c) 1999. Release: 01.06 ----!
!---- ----!
!---- Fast Fourier Transform Information ----!
!---------------------------------------------------------------------------!
Type: Differ. (Fo-Fc)
Grid Dimensions: Nx = 22 Dx = 0.045
Ny = 46 Dy = 0.022
Nz = 38 Dz = 0.026
Reflections used: 1269
H max = 6 H min = 0
K max = 14 K min = 0
L max = 16 L min = -11
F000 = 0.000 Scale = 1.000
Density Max: 2.402 Density Min: -1.943
!---------------------------------------------------------------------------!
!---- FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ----!
!---- Copyright(c) 1999. Release: 01.06 ----!
!---- ----!
!---- Scan Peaks Information ----!
!---------------------------------------------------------------------------!
Number of Peaks = 15
Peak x/a y/b z/c Occ Density
1 0.7764 0.1856 0.9667 1.0000 1.5709
2 0.4427 0.0316 0.8370 1.0000 1.6596
3 0.0877 0.0045 0.2073 1.0000 1.5802
4 0.6061 0.1012 0.6071 1.0000 1.5259
5 0.2309 0.1736 0.0937 1.0000 1.4568
6 0.7223 0.0691 0.3242 1.0000 1.3538
7 0.4581 0.2493 0.2926 1.0000 1.3787
8 0.2839 0.1734 0.0340 1.0000 1.2607
9 0.8174 0.1586 0.3924 1.0000 -1.4434
10 0.9348 0.2405 0.7604 1.0000 1.2146
11 0.7436 0.0188 0.4811 1.0000 -1.0543
12 0.2861 0.2462 0.4682 1.0000 -1.1870
13 0.1125 0.1226 0.5078 1.0000 1.2901
14 0.1478 0.0839 0.0706 1.0000 -1.0437
15 0.2733 0.1270 0.9614 1.0000 -0.9006
!---------------------------------------------------------------------------!
!---- FOURIER MAPS CALCULATION PROGRAM (Version: Windows 9X / NT) ----!
!---- Copyright(c) 1999. Release: 01.06 ----!
!---- ----!
!---- List Control of Atom / Peaks ----!
!---------------------------------------------------------------------------!
Atom x/a y/b z/c Peak x/a y/b z/c Dist
PB1 0.2194 0.1891 0.0553 5 0.2309 0.1736 0.0937 0.411
PB2 0.2384 0.5604 0.1340 6 0.7223 0.0691 0.3242 0.461
Cpu Time: 1.12 secs
Fourier Program finished OK
!***************************************************************************!
!**** E:\sdpd-co Date: 14/03/2000 Time: 09:47:15 ****!
!**** ****!
!**** Programme d'Actions Integrees Franco-Espagnol << PICASSO 1999 >> ****!
!**** (Dossier Num. 98186) ****!
!**** ****!
!***************************************************************************!
GFOUR is already saying that peaks 5 and 6 are too close from the Pb atoms.
Let us see if the positive peaks are really corresponding to F atoms
by using STRUVIR/STRUPLO :
TITL "PbF2/CrF3/HFaq" ??? P21/c CELL 5.463 11.208 9.574 90.00 91.20 90.00 PARA 1.00 1 1 1 1 1 1 3.20 0.00 0.01 SYMM +X,1/2-Y,1/2+Z VIEW 0 0 1 0 0 0 XYZR -0.50 1.50 -0.50 1.50 -0.50 1.50 TETR 109.0 20.0 1.62 0.20 OCTA 90.0 20.0 1.97 0.40 PERS 70.00 COLO PB 2 CR 3 F 4 Q 5 CIRC 0.5 PB END PB 1 0.2195 0.1891 0.0553 PB 2 0.2383 0.5604 0.1340 CR 1 0.2946 0.8675 0.2046 Q 1 0.7764 0.1856 0.9667 1.0000 1.5709 Q 2 0.4427 0.0316 0.8370 1.0000 1.6596 Q 3 0.0877 0.0045 0.2073 1.0000 1.5802 Q 4 0.6061 0.1012 0.6071 1.0000 1.5259 Q 5 0.2309 0.1736 0.0937 1.0000 1.4568 Q 6 0.7223 0.0691 0.3242 1.0000 1.3538 Q 7 0.4581 0.2493 0.2926 1.0000 1.3787 Q 8 0.2839 0.1734 0.0340 1.0000 1.2607 Q 10 0.9348 0.2405 0.7604 1.0000 1.2146 Q 13 0.1125 0.1226 0.5078 1.0000 1.2901
The distance calculation (in STRUVIR but not in STRUPLO) shows that
peaks 5 and 8 are not the only peaks too close to the Pb atoms. Peak 6
should also be eliminated. Then it is clear that peaks 1, 2, 3, 4, 7 and
10 will form a CrF6 octahedra. And peak 13 is a good candidate for being
an isolated F atom, connected only to the Pb atoms.
Just an example in order to show that you will not always obtain so easily very small reliability factors...
The first thing to do with that pattern, which you would not have any problem to identify by using the JCPDS-ICDD database, is to use the ICSD data for building a .pcr file for structure factor extraction purpose. But in fact, we just want to see how small can be the Rp and Rwp reliability factors when there is no structure constraint, but only cell constraint. This is always a good thing to do, in any case : verifying the cell, the space group, and having an idea of the limit of refinability of the pattern.
The ICSD file is below and seems complete :
COL ICSD Collection Code 24659 DATE Recorded Oct 3, 1986; updated Oct 3, 1986 NAME Potassium hexafluorotitanate FORM K2 Ti F6 = F6 K2 Ti TITL The Crystal Structure of K2 Ti F6 REF Acta Crystallographica (1,1948-23,1967) ACCRA 5 (1952) 683-684 AUT Siegel S CELL a=5.715(2) b=5.715(2) c=4.656(1) alpha=90.0 beta=90.0 gamma=120.0 V=131.7 D=3.07 Z=1 SGR P -3 m 1 (164) - trigonal CLAS -3m (Hermann-Mauguin) - D3d (Schoenflies) PRS hP9 ANX AB2X6 PARM Atom__No OxStat Wyck -----X----- -----Y----- -----Z----- -SOF- K 1 1.000 2d 1/3 2/3 0.700(4) Ti 1 4.000 1a 0. 0. 0. F 1 -1.000 6i 0.156(3) 0.844(3) 0.244(4) WYCK i d a TEST Calculated density unusual but tolerable. (Code 23) TEST No R value given in the paper. (Code 51) TEST At least one temperature factor missing in the paper. (Code 53)After evaluating manually the background (with DMPLOT), the .pcr for Le Bail fit is built. We use here the zeropoint and cell parameters refined by ERACEL, from peak positions estimated by PowderX and compared to the expected positions generated by HKLGEN :
COMM K2TiF6 P-3m1 ! Files => DAT-file: k2tif60, PCR-file: k2tif60 !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 1 20 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 0 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 20.0000 0.7995 0.0000 90.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 20 0.01 0.40 0.40 0.40 0.80 5.0000 0.0200 130.0000 0.000 0.000 ! ! 2Theta/TOF Background 5.000 4104.130 6.050 3431.700 8.110 2481.030 10.590 1889.760 12.870 1356.450 14.730 1059.970 16.190 864.180 19.910 614.380 23.230 523.230 29.010 421.960 33.770 391.580 47.430 364.580 60.690 334.190 74.970 303.810 95.450 256.550 100.010 256.550 104.350 256.550 109.530 300.440 127.730 388.210 130.000 398.330 ! ! Excluded regions (LowT HighT) 5.00 16.00 130.00 170.00 ! ! 1 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE -0.0810 11.00 0.0000 0.00 0.0000 0.00 0.000000 0.00 0 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 1.62 !------------------------------------------------------------------------------- K2TiF6 ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 0 0 0 0.0 0.0 1.0 2 0 0 0 0 0.00 0 5 0 ! P -3 M 1 <--Space group symbol ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.10000E-01 0.2989 0.0000 0.0000 0.0000 0.0000 0 0.00000 31.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.05000 -0.02500 0.02500 0.00000 0.00000 0.00000 0.00000 0 61.00 51.00 41.00 101.00 0.00 0.00 0.00 ! a b c alpha beta gamma 5.733000 5.733000 4.664600 90.000000 90.000000 120.000000 71.00000 71.00000 81.00000 0.00000 0.00000 71.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.12870 0.02391 0.00000 0.00000 0.00 0.00 91.00 21.00 0.00 0.00As usual, only the zeropoint is refined for a few cycles, and then new cycles are made, refining all reasonable parameters taking account of the data quality (2-theta-max = 130° allows refining U and W and even X). Below is the final .sum file :
******************************************************** ** PROGRAM FULLPROF.98 (Version 0.2 - Mar98-LLB JRC) ** ******************************************************** Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 14/03/2000 Time: 14:38:59.400 => PCR file code: k2tif60 => DAT file code: k2tif60 => Title: K2TiF6 P-3m1 ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 => Base of peaks: 2.0*HW* 20.00 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 130.0000 Step: 0.0200 No. of points: 6251 => Pattern Matching (fixed scale) for phase: 1 => Scor: 4.5072 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 10 ------------------------------------------------------------------------------ => Phase No. 1 K2TiF6 P -3 M 1 ------------------------------------------------------------------------------ => No. of reflections: 226 ==> PROFILE PARAMETERS: => Cell parameters : 5.73305 0.00007 5.73305 0.00007 4.66459 0.00006 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => overall scale factor : 0.010000000 0.000000000 => Eta(p-v) or m(p-vii) : 0.19519 0.01013 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.04474 0.00243 -0.02713 0.00234 0.02923 0.00057 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.14189 0.00289 0.03388 0.00082 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00799 0.00025 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: -0.0875 0.0008 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 20 => MaxCycle: 20 => N-P+C: 5689 => Rp: 6.47 Rwp: 9.77 Rexp: 2.79 Chi2: 12.3 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 8.94 Rwp: 12.2 Rexp: 3.47 Chi2: 12.3 => Deviance: 0.704E+05 Dev* : 12.37 => DW-Stat.: 0.2481 DW-exp: 1.9213 => N-sigma of the GoF: 601.700 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 5654 => Rp: 6.48 Rwp: 9.78 Rexp: 2.78 Chi2: 12.4 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 8.93 Rwp: 12.2 Rexp: 3.46 Chi2: 12.4 => Deviance: 0.704E+05 Dev* : 12.44 => DW-Stat.: 0.2482 DW-exp: 1.9211 => N-sigma of the GoF: 603.664 => Phase: 1 => Bragg R-factor: 1.63 Vol: 132.775( 0.003) Fract(%): 0.00( 0.00) => Rf-factor=0.971 ATZ: 0.000 Brindley: 1.0000 => Run finished at: Date: 14/03/2000 Time: 14:39:49.710Rp=8.9% and Rwp= 12.2 is not too bad. People say that chi2 must be close to 1.0. This is mainly a view of statistician, but beleve me, first trust Rp and Rwp. Suppose you just divide your intensity by a factor 2. Then your Rexp value will increase a lot, and your chi2 will decrease since chi=Rwp/Rexp, (chi2=chi**2) and Rwp will not be very different from the current value excepted if the noise becomes really more important on that new scaled pattern. Let us see the fit drawing :
A very nice fit, though the FWHM is large (between 0.15 and 0.37° 3-theta, due to the choice of a large 0.15° detector slit aperture on a Siemens D500, Bragg-Brentano, with graphite back monochromator). If there is a problem on that pattern, it will not be anisotropic broadening.
Let us now introduce the atoms with their coordinates in a new .pcr
file, and refine. Below is the final .sum file :
******************************************************** ** PROGRAM FULLPROF.98 (Version 0.2 - Mar98-LLB JRC) ** ******************************************************** Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 14/03/2000 Time: 15:40:33.300 => PCR file code: k2tif6 => DAT file code: k2tif6 => Title: K2TiF6 P-3m1 ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 => Base of peaks: 2.0*HW* 20.00 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 130.0000 Step: 0.0200 No. of points: 6251 => Crystal Structure Refinement for phase: 1 => Scor: 5.5089 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 17 ------------------------------------------------------------------------------ => Phase No. 1 K2TiF6 P -3 M 1 ------------------------------------------------------------------------------ => No. of reflections: 228 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. K 0.33333( 0) 0.66667( 0) 0.70649( 72) 2.659( 95) 0.200( 0) TI 0.00000( 0) 0.00000( 0) 0.00000( 0) 0.710( 65) 0.100( 0) F 0.15706( 26) 0.84294( 26) 0.19363( 73) 0.871( 88) 0.600( 0) ==> PROFILE PARAMETERS: => Cell parameters : 5.73158 0.00025 5.73158 0.00025 4.66423 0.00024 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => overall scale factor : 0.051430602 0.000433081 => Eta(p-v) or m(p-vii) : 0.17984 0.02985 => Overall tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.02734 0.00655 -0.00534 0.00645 0.02465 0.00158 => Preferred orientation: 1.00000 0.00000 0.00000 0.00000 => Asymmetry parameters : 0.08863 0.01435 0.03341 0.00254 0.00000 0.00000 0.00000 0.00000 => X and y parameters : 0.00874 0.00073 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: -0.1038 0.0037 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 8 => MaxCycle: 8 => N-P+C: 5682 => Rp: 18.3 Rwp: 26.1 Rexp: 2.79 Chi2: 87.9 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 25.3 Rwp: 32.5 Rexp: 3.47 Chi2: 87.9 => Deviance: 0.910E+06 Dev* : 160.1 => DW-Stat.: 0.0443 DW-exp: 1.9238 => N-sigma of the GoF: 4632.408 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 5640 => Rp: 18.3 Rwp: 26.2 Rexp: 2.78 Chi2: 88.6 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 25.2 Rwp: 32.5 Rexp: 3.46 Chi2: 88.6 => Deviance: 0.910E+06 Dev* : 161.3 => DW-Stat.: 0.0443 DW-exp: 1.9235 => N-sigma of the GoF: 4649.726 => Phase: 1 => Bragg R-factor: 21.1 Vol: 132.697( 0.011) Fract(%): 0.00( 0.00) => Rf-factor= 14.9 ATZ: 0.000 Brindley: 1.0000 => Run finished at: Date: 14/03/2000 Time: 15:41:03.400This is not really satisfying. Very high Rp, Rwp and Bragg R factors are obtained. The fit drawing shows that a few reflections have big intensity problems :
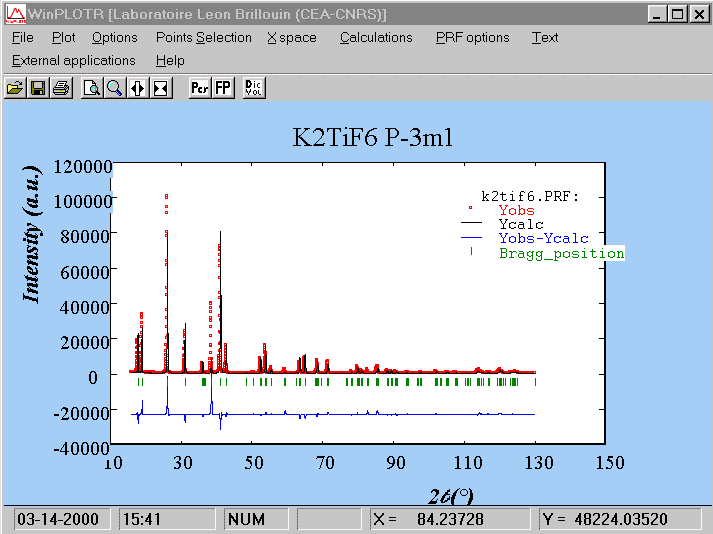
We may look at their hkl Miller indices in the .out file :
No. Code H K L Mult Hw ETA/M 2theta/TOF Icalc Iobs Sigma Square-StrFactor 1 1 0 1 0 3 0.156 0.336 17.855 1451.9 1301.4 4.542 65.4033 2 1 1 0 0 3 0.156 0.336 17.855 1451.9 1301.4 4.542 65.4032 3 1 0 0 1 2 0.157 0.346 19.011 3357.4 4114.7 7.361 257.4994 4 1 0 1 1 6 0.158 0.409 26.196 831.8 1014.9 1.884 41.2810 5 1 1 0 1 6 0.158 0.409 26.196 10548.2 12870.1 6.711 523.4990 6 1 1 1 0 6 0.159 0.452 31.184 4316.4 3405.1 6.152 309.2498 7 1 0 2 0 3 0.161 0.496 36.163 556.8 461.8 4.854 109.4788 8 1 2 0 0 3 0.161 0.496 36.163 556.8 461.8 4.854 109.4788 9 1 1 1 1 12 0.161 0.501 36.778 110.0 94.0 6.645 5.6073 10 1 0 0 2 2 0.162 0.517 38.573 1323.3 5608.3 12.526 448.6279 11 1 0 2 1 6 0.163 0.539 41.166 9975.3 9082.4 4.972 1298.2241 12 1 2 0 1 6 0.163 0.539 41.166 3910.0 3560.0 3.113 508.8614 13 1 0 1 2 6 0.164 0.554 42.807 2820.0 2798.4 6.477 399.6232 14 1 1 0 2 6 0.164 0.554 42.807 11.0 10.9 0.405 1.5628 15 1 1 2 0 6 0.167 0.603 48.482 32.8 12.1 11.956 6.0994 16 1 2 1 0 6 0.167 0.603 48.482 32.8 12.1 11.956 6.0994 17 1 1 1 2 12 0.168 0.620 50.407 56.4 120.7 12.862 5.7194 18 1 1 2 1 12 0.169 0.639 52.533 329.5 309.3 2.812 36.5478 19 1 2 1 1 12 0.169 0.639 52.533 1457.9 1368.7 5.915 161.7158 20 1 0 2 2 6 0.170 0.651 53.903 2356.0 2428.4 5.562 552.8826 21 1 2 0 2 6 0.170 0.651 53.903 842.7 868.6 3.327 197.7646 22 1 0 3 0 3 0.171 0.665 55.491 303.6 338.6 5.345 151.7907 23 1 3 0 0 3 0.171 0.665 55.491 303.6 338.6 5.345 151.7907 24 1 0 3 1 6 0.174 0.697 59.204 129.3 289.6 12.359 37.1657 25 1 3 0 1 6 0.174 0.697 59.204 5.4 12.0 2.518 1.5424 26 1 0 0 3 2 0.175 0.699 59.397 3.1 21.7 4.221 2.6904 27 1 0 1 3 6 0.177 0.726 62.554 54.1 31.1 5.371 17.4700 28 1 1 0 3 6 0.177 0.726 62.554 35.5 20.4 4.352 11.4661 29 1 1 2 2 12 0.178 0.735 63.598 1948.7 1560.6 6.110 325.5098 30 1 2 1 2 12 0.178 0.735 63.598 51.3 41.1 0.992 8.5749 31 1 2 2 0 6 0.180 0.748 65.037 2456.2 1986.5 5.927 859.0827 32 1 1 3 0 6 0.183 0.774 68.045 68.1 52.6 3.953 26.0845 33 1 3 1 0 6 0.183 0.774 68.045 68.1 52.6 3.953 26.0845 34 1 2 2 1 12 0.183 0.778 68.439 384.6 301.0 3.873 74.4566 35 1 1 1 3 12 0.184 0.779 68.618 1378.8 1372.2 5.740 268.3156 36 1 0 3 2 6 0.185 0.788 69.614 57.1 42.3 6.343 22.8403 37 1 3 0 2 6 0.185 0.788 69.614 1.3 1.0 0.955 0.5179 38 1 1 3 1 12 0.187 0.803 71.378 738.8 774.4 5.870 154.9543 39 1 3 1 1 12 0.187 0.803 71.378 81.8 85.7 1.953 17.1493 40 1 0 2 3 6 0.187 0.805 71.553 3.8 4.3 0.380 1.5978 41 1 2 0 3 6 0.187 0.805 71.553 841.3 961.8 5.660 354.5250 42 1 0 4 0 3 0.194 0.850 76.739 12.3 10.4 10.265 11.6689 43 1 4 0 0 3 0.194 0.850 76.739 12.3 10.4 10.265 11.6689 44 1 2 2 2 12 0.196 0.863 78.238 305.7 282.3 8.363 75.0521 45 1 0 4 1 6 0.198 0.878 79.931 180.2 205.7 3.164 91.3770 46 1 4 0 1 6 0.198 0.878 79.931 724.0 826.8 6.343 367.2465 47 1 1 2 3 12 0.199 0.880 80.099 102.6 106.9 2.328 26.1090 48 1 2 1 3 12 0.199 0.880 80.099 2.4 2.5 0.357 0.6150 49 1 1 3 2 12 0.200 0.888 81.043 20.8 21.6 1.328 5.3731 50 1 3 1 2 12 0.200 0.888 81.043 506.7 527.6 6.560 131.0646 51 1 0 0 4 2 0.203 0.902 82.689 66.4 471.9 17.226 105.9778 52 1 2 3 0 6 0.207 0.924 85.130 83.3 117.4 4.804 45.9082 53 1 3 2 0 6 0.207 0.924 85.130 83.3 117.4 4.804 45.9082 54 1 0 1 4 6 0.208 0.926 85.464 17.9 36.7 1.654 9.9255The biggest errors seem to come from the 00l reflections, always observed larger than calculated. This can also be seen by using some drawing program like DMPLOT :
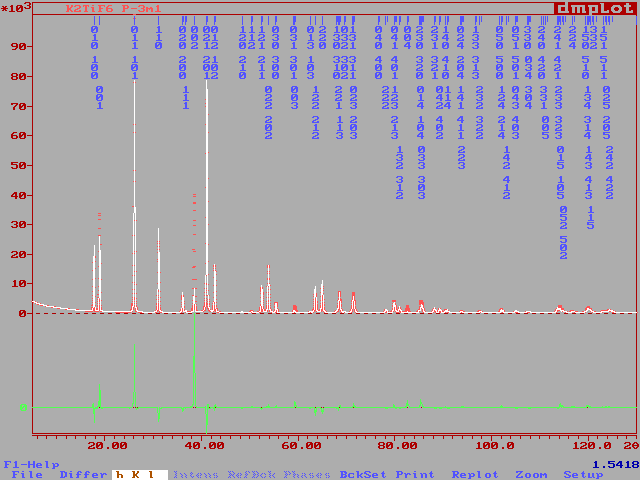
We may thus suppose that, in spite of the precaution used for the sample preparation (vertical load of the sample holder), there could be some preferred orientation along the 00l direction.
Then we may test the March-Dollase option for a correction :
COMM K2TiF6 P-3m1 ! Files => DAT-file: k2tif6, PCR-file: k2tif6 !Job Npr Nph Nba Nex Nsc Nor Dum Iwg Ilo Ias Res Ste Nre Cry Uni Cor 0 5 1 20 2 0 1 1 0 0 1 0 0 0 0 0 0 ! !Ipr Ppl Ioc Mat Pcr Ls1 Ls2 Ls3 Syo Prf Ins Rpa Sym Hkl Fou Sho Ana 0 0 1 0 1 0 0 0 1 1 0 1 0 0 2 0 0 ! ! lambda1 Lambda2 Ratio Bkpos Wdt Cthm muR AsyLim Rpolarz 1.540560 1.544390 0.5000 90.0000 20.0000 0.7995 0.0000 90.00 0.0000 ! !NCY Eps R_at R_an R_pr R_gl Thmin Step Thmax PSD Sent0 8 0.01 0.40 0.40 0.40 0.60 5.0000 0.0200 130.0000 0.000 0.000 ! ! 2Theta/TOF Background 5.010 4104.130 6.050 3431.700 8.110 2481.030 10.590 1889.760 12.870 1356.450 14.730 1059.970 16.190 864.180 19.910 614.380 23.230 523.230 29.010 421.960 33.770 391.580 47.430 364.580 60.690 334.190 74.970 303.810 95.450 256.550 100.010 256.550 104.350 256.550 109.530 300.440 127.730 388.210 130.010 398.330 ! ! Excluded regions (LowT HighT) 5.00 16.00 130.00 170.00 ! ! 18 !Number of refined parameters ! ! Zero Code Sycos Code Sysin Code Lambda Code MORE -0.1038 11.00 0.0000 0.00 0.0000 0.00 0.000000 0.00 0 !------------------------------------------------------------------------------- ! Data for PHASE number: 1 ==> Current R_Bragg: 21.10 !------------------------------------------------------------------------------- K2TiF6 ! !Nat Dis Mom Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More 3 0 0 0.0 0.0 1.0 0 0 0 0 0 0.00 0 5 0 ! P -3 M 1 <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t /Codes K K+1 0.33333 0.66667 0.70649 2.65903 0.20000 0 0 0 0.00 0.00 111.00 121.00 0.00 TI TI+4 0.00000 0.00000 0.00000 0.71041 0.10000 0 0 0 0.00 0.00 0.00 131.00 0.00 F F-1 0.15706 0.84294 0.19363 0.87125 0.60000 0 0 0 141.00 -141.00 151.00 161.00 0.00 ! Scale Shape1 Bov Str1 Str2 Str3 Strain-Model 0.51431E-01 0.1798 0.0000 0.0000 0.0000 0.0000 0 21.00000 31.00 0.00 0.00 0.00 0.00 ! U V W X Y GauSiz LorSiz Size-Model 0.02734 -0.00534 0.02465 0.00874 0.00000 0.00000 0.00000 0 61.00 51.00 41.00 171.00 0.00 0.00 0.00 ! a b c alpha beta gamma 5.731583 5.731583 4.664235 90.000000 90.000000 120.000000 71.00000 71.00000 81.00000 0.00000 0.00000 71.00000 ! Pref1 Pref2 Asy1 Asy2 Asy3 Asy4 1.00000 0.00000 0.08863 0.03341 0.00000 0.00000 181.00 0.00 91.00 101.00 0.00 0.00After a few cycles, there is a strong improvement in the fit, as seen in the .sum file :
******************************************************* *** PROGRAM FULLPROF (Version 3.3 - Aug97-LLB JRC) *** ******************************************************* Rietveld, Profile Matching & Integrated Intensity Refinement of X-ray and/or Neutron Data Date: 03/14/00 Time: 17:15:37.97 => PCR file code: k2tif6 => DAT file code: k2tif6 => Title: K2TiF6 P-3m1 ==> CONDITIONS OF THIS RUN: => Global Refinement of X-ray powder diffraction data Bragg-Brentano or Debye-Scherrer geometry => The 5th default profile function was selected => Data supplied in free format => Wavelengths: 1.54056 1.54439 => Base of peaks: 2.0*HW* 20.00 => Cos(Monochromator angle)= 0.7995 => Absorption correction (muR-eff): 0.0000 ==> Angular range, step and number of points: 2Thmin: 5.0000 2Thmax: 130.0000 Step: 0.0200 No. of points: 6251 => Crystal Structure Refinement for phase: 1 => Scor: 5.3827 ==> RESULTS OF REFINEMENT: => No. of fitted parameters: 18 ------------------------------------------------------------------------------ => Phase No. 1 K2TiF6 P -3 M 1 ------------------------------------------------------------------------------ => No. of reflections: 228 ==> ATOM PARAMETERS: Name x sx y sy z sz B sB occ. socc. K 0.33333( 0) 0.66667( 0) 0.70355( 30) 1.777( 51) 0.200( 0) TI 0.00000( 0) 0.00000( 0) 0.00000( 0) 1.661( 60) 0.100( 0) F 0.15129( 20) 0.84871( 20) 0.22209( 32) 1.379( 57) 0.600( 0) ==> PROFILE PARAMETERS: => Cell parameters : 5.73262 0.00016 5.73262 0.00016 4.66423 0.00013 90.00000 0.00000 90.00000 0.00000 120.00000 0.00000 => Overall scale factor : 0.055830561 0.000288385 => ETA(p-V) or M(P-VII) : 0.12428 0.01841 => Overall Tem. factor : 0.00000 0.00000 => Halfwidth parameters : 0.01682 0.00359 0.00120 0.00377 0.02348 0.00097 => Preferred orientation: 0.73477 0.00178 0.00000 0.00000 => Asymmetry parameters : 0.10102 0.00899 0.03409 0.00159 0.00000 0.00000 0.00000 0.00000 => X and Y parameters : 0.00990 0.00046 0.00000 0.00000 => Strain parameters : 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 => Size parameters : 0.00000 0.00000 0.00000 0.00000 ==> GLOBAL PARAMETERS: => Zero-point: -0.0977 0.0023 => Cos( theta)-shift parameter : 0.0000 0.0000 => Sin(2theta)-shift parameter : 0.0000 0.0000 ==> RELIABILITY FACTORS WITH ALL NON-EXCLUDED POINTS: => Cycle: 8 => MaxCycle: 8 => N-P+C: 5681 => Rp: 12.8 Rwp: 16.7 Rexp: 2.79 Chi2: 35.8 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 17.7 Rwp: 20.8 Rexp: 3.47 Chi2: 35.8 => Deviance: 0.213E+06 Dev* : 37.52 => DW-Stat.: 0.0922 DW-exp: 1.9241 => N-sigma of the GoF: 1854.572 ==> RELIABILITY FACTORS FOR POINTS WITH BRAGG CONTRIBUTIONS: => N-P+C: 5638 => Rp: 12.8 Rwp: 16.7 Rexp: 2.78 Chi2: 36.1 L.S. refinement => Conventional Rietveld R-factors ==> => Rp: 17.6 Rwp: 20.8 Rexp: 3.46 Chi2: 36.1 => Deviance: 0.213E+06 Dev* : 37.80 => DW-Stat.: 0.0922 DW-exp: 1.9239 => N-sigma of the GoF: 1861.713 => Phase: 1 => Bragg R-factor: 12.8 Vol: 132.745 Fract(%): 0.00 => Rf-factor= 10.4 ATZ: 0.000 Brindley: 1.0000However, a Bragg R factor of 12.8 % is still unacceptable, and the Rp value of 17.6 % is too much far from the value obtained without the structure contraint (< 9%).
The preferred orientation parameter is equal to 0.73 (instead of 1.00 if no preferred orientation occured). This is huge !
We may consider that it will not be possible to treat correctly such data, and record a new pattern more carefully. Possibly by the technique of dusting the sample through a sieve on the sample holder, or using some spray dry technique. This is the best thing to do, anyway, before to waste time by trying anisotropic thermal parameters, or disorder, or anything else.
This shows the limits of a preferred orientation correction by the March Dollase option in a Rietveld program.
Final plot as seen by the DMPLOT program. Some people may found that fit acceptable, but it is certainly not :
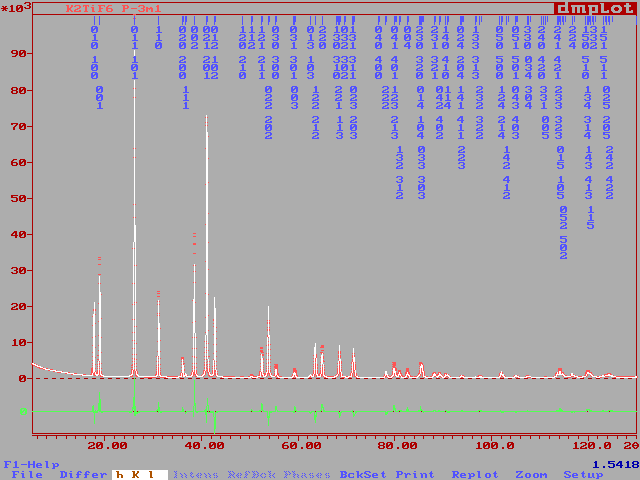
STRATEGY - SUMMARY - CHECKLIST
We hope that you have now an idea of the strategies commonly selected
for performing a Rietveld refinement (with structure constraint - note
that the strategy is different when there is only a cell contraint like
in the Pawley or Le Bail methods) :