Results for Sample 1
Indexing could be obtained from either TREOR, ITO or DICVOL.
Nothing was found when searching in the JCPDS-ICDD data base nor in ICSD. Due to clear strong preferred orientation effects on patterns zhu1 and zhu2, the zhu3 pattern was selected for structure factor extraction. The background was estimated manually with the DMPLOT program. The program FULLPROF was used (Le Bail method) selecting P21/m space group (which is unimportant at this stage). Links are given to files renamed as .txt files :
R-Factors (%) were : Rp = 4.65, Rwp = 6.33 (background included) and Rp = 7.59, Rwp = 9.41 (background subtracted). The fit, as shown by DMPLOT, seems acceptable.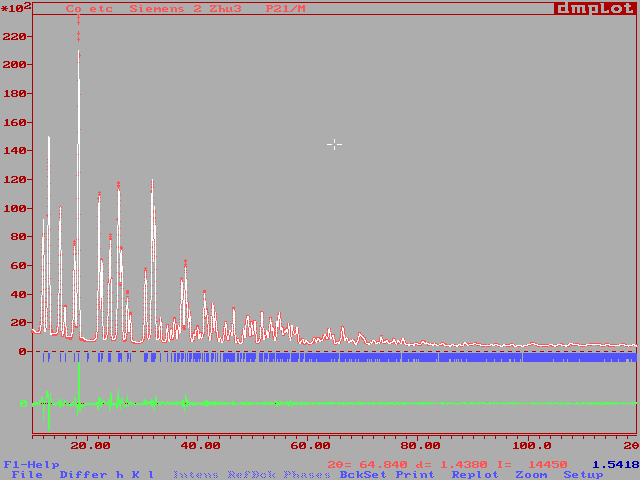{kind=link}
Six data files were prepared for use by SHELXS-97. The first data contains the whole data, with 780 hkl, (zhu0.hkl) and the OVERLAP program eliminated reflections having a neighbouring one at less than :
- 0.01° (2-theta) 629 hkl : zhu1.hkl
- 0.02° (2-theta) 526 hkl : zhu2.hkl
- 0.03° (2-theta) 434 hkl : zhu3.hkl
- 0.04° (2-theta) 355 hkl : zhu4.hkl
- 0.05° (2-theta) 295 hkl : zhu5.hkl
- P21/m : zhu0.lst ; zhu1.lst ; zhu2.lst ; zhu3.lst ; zhu4.lst ; zhu5.lst
- P21 : zhunc0.lst ; zhunc1.lst ; zhunc2.lst ; zhunc3.lst ; zhunc4.lst ; zhunc5.lst
The zhu2, zhunc2, zhunc3 and zhunc4 SHELXS-97 output give all the same proposition for the Co coordinates. The P21/m spacegroup could not lead to any coherent result. None of the 3 above propositions in the P21 space group led to the complete structure (remember the FWHM minimal is 0.25° 2-theta !). However, using the zhu5.hkl as reduced data set for testing the 3 propositions, it was possible to obtain R ~ 30% (with SHELX-76), keeping ~ten of the proposed atoms. Then, several cycles of Rietveld refinement and Fourier syntheses, revealed the whole structure. The final refinement was made either with or without soft constraints. Hydrogen atoms were not located. At least the positions of the water hydrogen atoms may be guessed from an evident O-H...O bonding scheme.
To be noted is the anisotropic broadening which affects particularly the patterns with best resolution (zhu1.txt and zhu2.txt). The 0k0 reflections are the narrower and were fitted separately by introducing a pseudo two-phases system :
The preferred orientation in zhu1 and zhu2 patterns seems to correspond to several orientations including 100 and 010. The best fit with structure constraint (although with problems), is obtained from the choice of the 001 direction leading to a March-Dollase parameter value >> 1 (indicating that the preferred orientation is all but the 001 one, may be the microcrystal shape correspond to needles with 001 as long axis). The zhu1 and zhu2 patterns were provided for possible use by methods able to cope with preferred orientation, and even to take advantage of it (their resolution is two times better than that of the zhu3 pattern).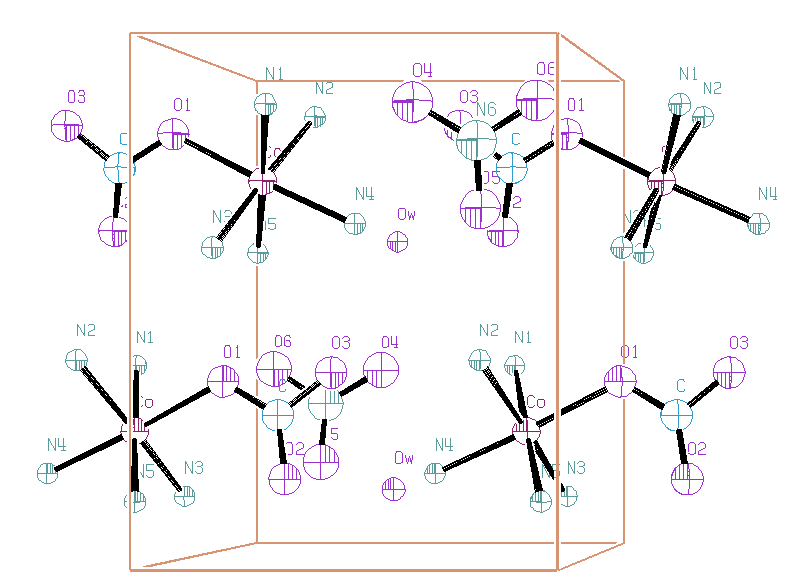{kind=link}
Thanks for your participation.
Copyright © 1998 L.M.D Cranswick & A. Le Bail