SDPD Internet Course
Week 6
Structure solution by Patterson, direct or molecule location methods, part 1 : Conventional methods.
Lectures
Read chapter 3.3 of the SDPD tutorial (CD) or/and access to the Kunming Workshop (CD) conference text, same chapter (middle of part 3). Some examples of structure solution by classical approach are given. You should play with those data (Na2C2O4, [Pd(NH3)4]Cr2O7, t-AlF3, beta-BaAlF5 and cimetidine C10H16N6S) before to try to solve the exercice given below.
Have a look at the SDPD Round Robin 1 (1998) (CD) and 2 (2002) (CD). A series of problems is available with various solutions proposed by competitors on an international scale.
Read the book chapters :
B4 : Chapter 2, pages 61-137 and
Chapter 5, pages 319-397.
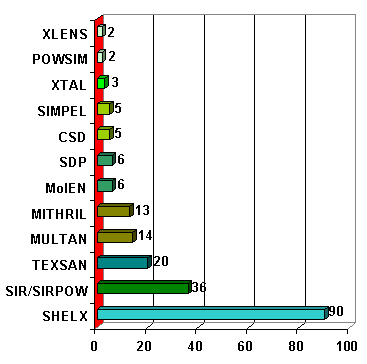
This week will be limited to the use of conventional methods, mostly by applying single-crystal derived software : mainly SHELXS for Patterson and/or direct methods. The (almost) complete list of software that were used for solving structures from powder diffraction data is given in the SDPD-Database (CD). We have not Internet links to all of them. Statistics place SHELXS at the head :
Software to download
At that time, you should already have obtained SHELX and WinGX from their authors, as well as SIR97. You may want to control SHELX by WinGX or another package. You may be interested in other software, like :
Caos by S. Cerrini & R. Spagna
Crunch by R. de Gelder and R.A.G. de Graaff (direct methods), only runs on Unix (and Linux). See the tutorial at CCP14 (CD).
Dirdif by P. T. Beurskens, Patterson and direct methods applied to difference structure factors. A MS Windows version, WinDirdif is available (CD).
MITHRIL by C. Gilmore, included in commercial package like TeXsan.
Jana by V. Petricek and M. Dusek, covers all usual tasks of the structure analysis except of plotting program and direct methods (CD).
Multan by P. Main, included in many commercial packages (?). You may download the Fortran source (CD) of an old (1980) version with the corresponding manual.
SDP commercial
SnB Shake and Bake
TeXsan commercial by MSC (Rigaku)
UNICS by Sakurai & Kobayashi
Xlens by J. Rius
etc.
Note that GSAS
performs Patterson analysis (CD).
Other software
Beside the above method software, some small programs may be useful in order to eliminate those "|Fobs|" that belong to reflections which are overlapping too much :
Overlap by Armel Le Bail (CD).
Some packages include special powder diffraction data treatment (Expo, Dorees in Powsim, FIPS, Focus....), this will be the next week subject, in fact limited by the unavailability (without co-signing a paper with the author ?) of some of these software. But if you find links, tell us please.
Structure viewing software
Visualizing complete or partial models is one of the keys for success. So that, you will need this week to download some software of this kind :
Ortep-3 for Windows by Louis Farrugia (CD : ortep32.zip - you have to ask for a licence to the author), based on Ortep-3 by Michael N. Burnett and Carroll K. Johnson
Struplo for Windows by Louis Farrugia (CD : struplo.zip - you have to ask for a licence to the author), based on Struvir (CD) by Armel Le Bail
Struvir by Armel Le Bail, open Fortran source DOS version (CD) based on Struplo by R. X. Fischer
Platon for Windows by Louis Farrugia (CD: pwt.zip), based on Platon by Ton Spek
Schakal was recently made available freely to the academic world (CD: Schakal.zip).
Chime based on Rasmol, plug-in for browser (CD : ch203w32.exe)
DrawXTL by Larry Finger and Martin Kroeker (CD : drawv31.exe)
Jamm by John N. Huffman
JSV by Steffen Weber
Ortex by Patrick McArdle
PowderCell by Werner Kraus and Gert Nolze (CD: pcw23.exe)
Rasmol by R. Sayle and Protein Explorer
WebLab viewer lite par Accelrys (CD : WLViewerLite40.exe)
Xtal-3D by M. hewat (VRML file builder)
Xtaldraw by K.L. Bartelmehs
See the CCP14 Web page on that subject (CD).
A tremendous list of commercial software offer you drawing possibility too (Atoms, Crystal Maker, Diamond, etc).
You will find VRML a cheap way to access to three dimensional views of your models. An old list of VRML tools for crystallography is available on this Web site (CD). Your browser should be equipped with a VRML viewer. like Cosmoplayer (CD : cosmo_win95nt_eng.exe).
Suggest us your favourite one, if it is
not listed here, thanks.
Exercise
The previous weeks, you extracted the "|Fobs|" for two unknown compounds synthesized in the systems NaF/CrF3 and PbF2/CrF3/HFaq.
Try now to obtain a model for these unknowns which would allow starting a refinement by the Rietveld method. But, at this stage, do not make use at all of the Rietveld method. Try to obtain the largest structure model from the "|Fobs|" (or of a partial dataset) by using Patterson and/or direct methods, and the single crystal structure refinement software of your choice. You can do Fourier difference synthesis, and try to locate the maximum of atoms. You may either use your own "|Fobs|" or the following ones in SHELX format (card HKLF 3) : na5.hkl and pbcr.hkl (the .fou Fullprof file renamed with a line of zero at the end and the text and phase 2 for pbcr removed). You may also need the Fullprof summary files corresponding to these structure factor extractions : na5.sum and pbcr.sum containing the final cell parameters, if you intend to use the overlap software. The structure completion by alternating Rietveld refinement/new "|Fobs|" extraction/Fourier syntheses, will be the subject of weeks 9-10 : so, do not do it now, we just want to see how far you can go with those first extracted "|Fobs|", because this is always the best thing to do before to try too soon to test the structure model by the Rietveld method. Do not use the Expo software this week, we want you to put your hands in the "garbage" (although most Patterson and direct methods software are "black boxes") before to try almost "automatic" software.
Who knows, you could determine already the whole structures from these "|Fobs|" ? As a result, we ask you for the atomic coordinates of all the atoms which you think may constitute the best starting model to enter into Rietveld refinements. Give also the space group and cell parameters. Explain in no more than 20 lines how you did the job.
Crystal chemistry knowledge is absolutely
necessary before starting a structure determination (but do not have too
much preconceived ideas). Cr3+ in fluorides is rarely in a coordination
different from 6 (octahedron), with Cr-F distances of the order of 1.9
Angstrom. Na+ may adopt 6-9 coordination in fluorides,
with Na-F ~2.2-2.9 Angstroms. Pb2+ may adopt 7-10 coordination,
with Pb-F ~ 2.4-3.0 Angstroms. Standard F-F distances could be 2.5-2.9
Angstroms.
Software selected for the correction
Fullprof for extracting "|Fobs|"; Overlap
for building reduced data set; SHEXLS-97 for Patterson and Direct methods;
Ortep-3 for Windows and Struplo for Windows for drawing and viewing; Cosmo
Player for VRML viewing; SHELXL-97 for structure refinement.
The next week will be reserved for Structure
solution by Patterson, direct or molecule location methods, part 2.
Good luck !